




























目 录
第一章 药品注册申请受理情况
(一)总体情况
(二)技术审评类药品注册申请受理情况
1. 中药注册申请受理情况
2. 化学药品注册申请受理情况
3. 生物制品注册申请受理情况
(三)直接行政审批类注册申请受理情况
第二章 药品注册申请审评审批情况
(一)总体情况
(二)技术审评类注册申请审结情况
1. 中药注册申请审结情况
2. 化学药品注册申请审结情况
3. 生物制品注册申请审结情况
(三)直接行政审批类注册申请审结情况
第三章 加快新药好药上市,满足临床患者需求
(一)突破性治疗药物程序
(二)附条件批准程序
(三)优先审评审批程序
第四章 药品研发与审评沟通交流情况
(一)沟通交流会议申请与办理情况
(二)一般性技术问题咨询情况
第五章 药品研发指导原则方面工作
(一)持续完善审评标准体系建设
(二)ICH 指南文件的转化实施
第六章 积极推动监管科学研究,服务行业高质量发展
(一)药品监管科学的总体情况
(二)药品监管科学项目的组织实施
(三)药品监管科学项目主要成效
第七章 药品研发与技术审评宣贯与培训
第八章 2023年度药品审评主要工作回顾
附件5 2023 年国家药监局批准的药品纳入加快上市程序情况
附件8 2023 年药审中心发布的指导原则
附件9 ICH 正在活跃的议题
附件10 2023 年药审中心开展的培训
第一章 药品注册申请受理情况
(一)总体情况
2023 年,药品注册申请申报量持续增长,药审中心受理各类注册申请 18503 件(同比增加 35.84%,以受理号计,下同),包括药品制剂注册申请16898 件(同比增加36.63%),化学原料药注册申请1605件(同比增加 28.09%)。16898 件药品制剂注册申请包括技术审评类注册申请 13153 件(同比增加 41.41%,包括 13144 件药品,9 件药械组合),直接行政审批类注册申请 3745 件(同比增加 22.11%,包括补充申请和一次性进口)。2019 年至2023 年注册申请受理情况详见图1。
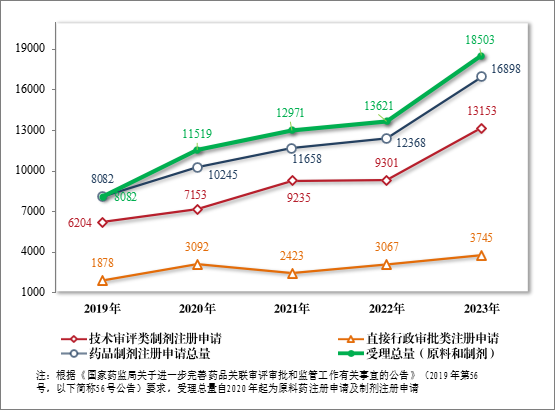
图 1 2019 年至 2023 年注册申请受理情况(件)
(二)技术审评类药品注册申请受理情况
2023 年受理的 13144 件技术审评类药品注册申请中,以药品类型统计,中药注册申请 1163 件,同比增加 176.25%;化学药品注册申请 9813 件,同比增加 39.11%,占全部需技术审评的药品注册申请受理量的 74.66%;生物制品注册申请 2168 件,同比增加 19.12%。2019年至 2023 年需技术审评的各药品类型注册申请受理情况详见图2。
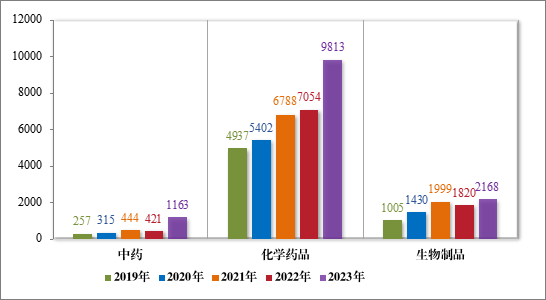
图 2 2019 年至 2023 年需技术审评的各药品类型注册申请受理情况(件)
以注册申请类别统计,受理新药临床试验申请(该注册申请类别以下简称“IND”)2997 件,同比增加 33.56%;验证性临床试验申请 170 件,比 2022 年增加 32.81%;新药上市许可申请(该注册申请类别以下简称“NDA”)470 件,同比增加 40.72%;同名同方药、化学仿制药上市许可申请(该注册申请类别以下简称“ANDA”)3852 件,同比增加 66.25%;仿制药质量和疗效一致性评价注册申请(该注册申请类别以下简称“一致性评价申请”)1006 件,同比增加 20.48%;补充申请 4115 件,同比增加 36.26%;境外生产药品再注册申请 534 件,同比增加 28.06%。2019 年至 2023 年需技术审评的各类别注册申请受理情况详见图 3。
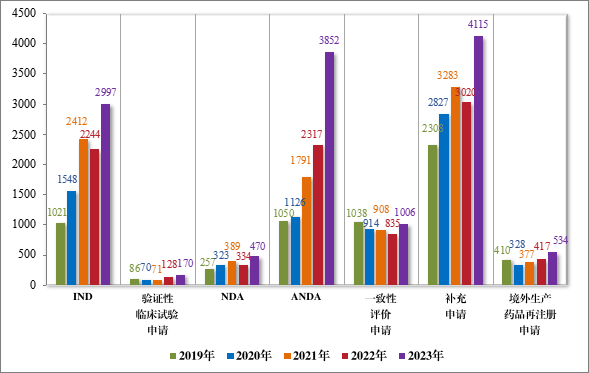
图 3 2019 年至 2023 年需技术审评的各类别注册申请受理情况(件)
1. 中药注册申请受理情况
2023 年受理中药注册申请1163 件。按审评序列统计,IND 75 件,同比增加 31.58%;NDA 26 件,同比增加 85.71%;补充申请 1054 件,同比增加 206.40%;ANDA 1 件,进口再注册 7 件,与去年持平。2019年至 2023 年需技术审评的中药各类别注册申请受理情况详见图 4。
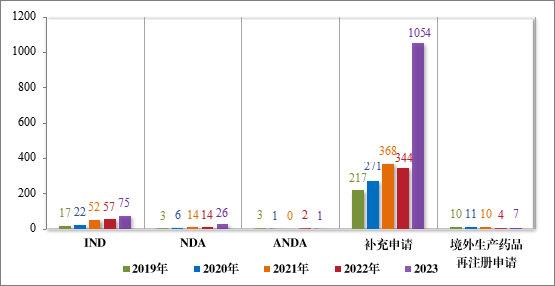
图 4 2019 年至 2023 年需技术审评的中药各类别注册申请受理情况(件)
以注册分类统计,IND 75 件,包括创新中药 IND 54 件(47 个品种),同比增加 38.46%;改良型中药 IND 21 件(21 个品种),同比增加 23.53%。NDA 26 件,包括创新中药 NDA 8 件(7 个品种),与去年持平;改良新中药 NDA3 件(3 个品种);古代经典名方中药复方制剂 NDA 15 件(11 个品种),同比增加 275.00%。
2023 年各注册分类中药 IND、NDA 受理情况详见表 1,2020 年至 2023 年各注册分类中药 IND 受理情况详见图 5,2020 年至 2023年各注册分类中药 NDA 受理情况详见图 6。
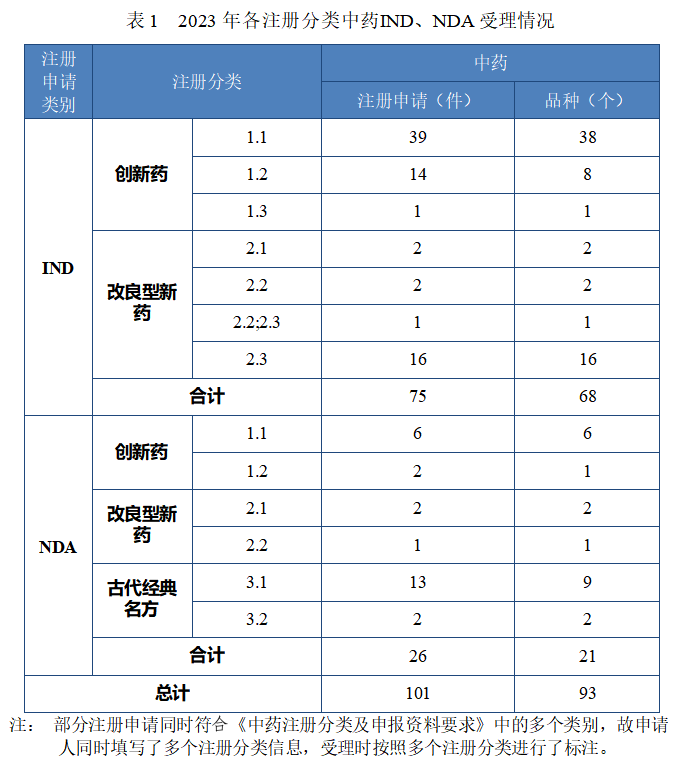
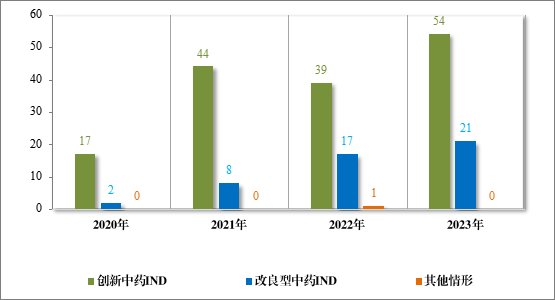
图 5 2020 年至 2023 年各注册分类中药IND 受理情况(件)
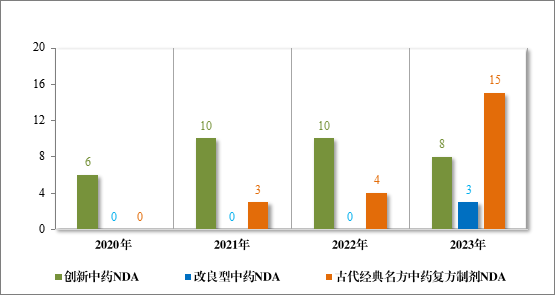
图 6 2020 年至 2023 年各注册分类中药NDA 受理情况(件)
2. 化学药品注册申请受理情况
2023 年受理需技术审评的化学药品注册申请 9813 件。按审评序 列统计,IND 1778 件,同比增加66.48%;NDA 249 件,同比增加31.75%; ANDA 3851 件,同比增加 66.35%;一致性评价申请 1006 件,同比增加 20.48%。5.1 类化学药品1注册申请 130 件,同比减少 10.96%,其中验证性临床试验申请2共 32 件,NDA 98 件。2019 年至 2023 年需技术审评的化学药品各类别注册申请受理情况详见图 7。
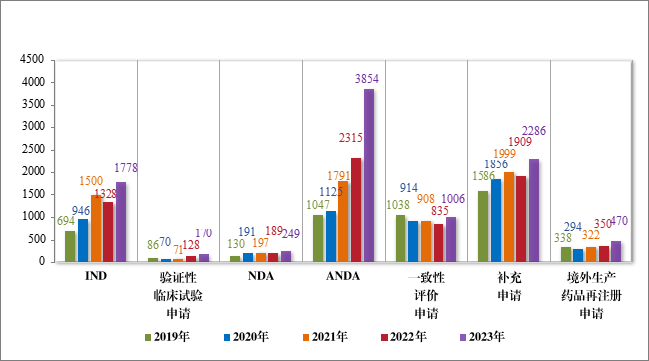
图 7 2019 年至 2023 年需技术审评的化学药品各类别注册申请受理量(件)
1778 件 IND 中,包括创新化学药品 IND 1368 件(600 个品种),同比增加 30.78%;改良型化学药品 IND 410 件(229 个品种),同比增加 45.39%。NDA 249 件中,包括创新化学药品 NDA 79 件(55 个品种),同比增加 172.41%;改良型化学药品 NDA 72 件(46 个品种),同比增加 35.85%;5.1 类化学药品 NDA 98 件(62 个品种),同比减少 8.41%。
2023 年各注册分类化学药品 IND、NDA 受理情况详见表 2,2019年至 2023 年各注册分类化学药品临床试验申请受理情况详见图 8, 2019 年至 2023 年各注册分类化学药品 NDA 受理情况详见图 9。
1 5.1 类化学药品为境外上市的原研药品和改良型药品的境内上市。
2 临床试验申请包括 IND 和验证性临床试验申请。

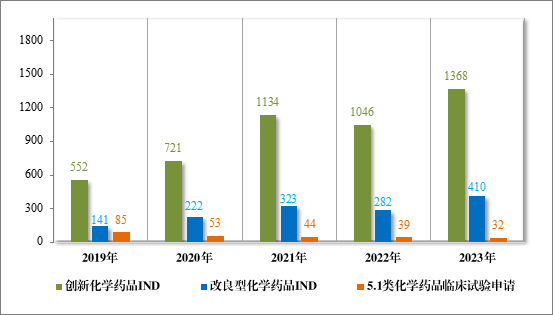
图 8 2019 年至 2023 年各注册分类化学药品临床试验申请受理情况(件)
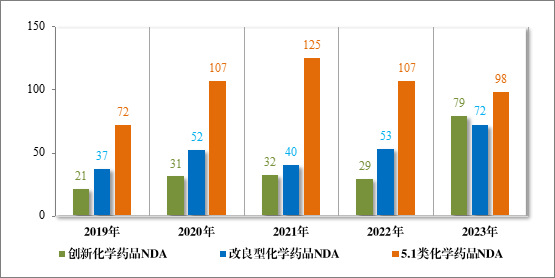
图 9 2019 年至 2023 年各注册分类化学药品NDA 受理情况(件)
3. 生物制品注册申请受理情况
2023 年受理生物制品注册申请 2168 件,其中,预防用生物制品注册申请 189 件、治疗用生物制品注册申请 1969 件和体外诊断试剂10 件。
以注册申请类别统计,IND 1144 件,同比增加 33.18%;NDA 195件,同比增加 48.85%;补充申请 772 件,同比增加 0.65%;境外生产药品再注册申请 57 件,同比减少 9.52%。2019 年至 2023 年需技术审评的生物制品各类别注册申请受理情况详见图10。
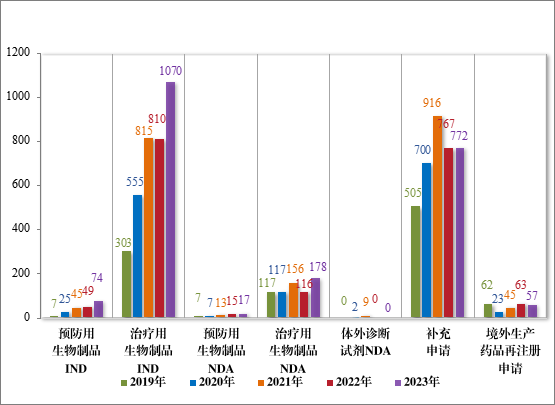
图 10 2019 年至 2023 年需技术审评的生物制品各类别注册申请受理情况(件)
以注册分类统计,预防用生物制品 IND 74 件,包括创新预防用生物制品 IND 43 件(34 个品种),同比增加 104.76% ;改良型预防用生物制品 IND13 件(11 个品种),同比减少 18.75% ;境内或境外已上市预防用生物制品 IND 18 件(11 个品种),同比增加 50.00% 。预防用生物制品 NDA 17 件,包括创新预防用生物制品 NDA 1 件(1个品种);改良型预防用生物制品 NDA 2 件(2 个品种);境内或境外已上市预防用生物制品 NDA 14 件(6 个品种)。
2023 年各注册分类预防用生物制品 IND、NDA 受理情况详见表3,2020 年至 2023 年各注册分类预防用生物制品 IND 受理情况详见图 11,2020 年至 2023 年各注册分类预防用生物制品 NDA 受理情况详见图 12。
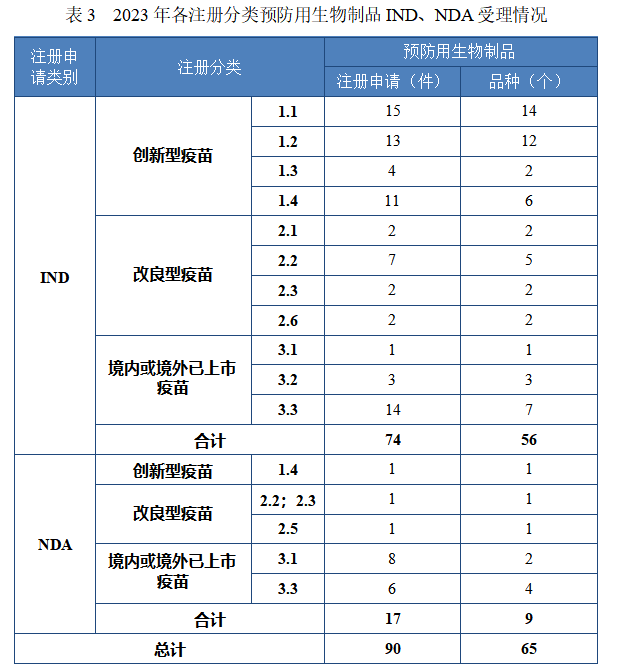

图 11 2020 年至 2023 年各注册分类预防用生物制品IND 受理情况(件)
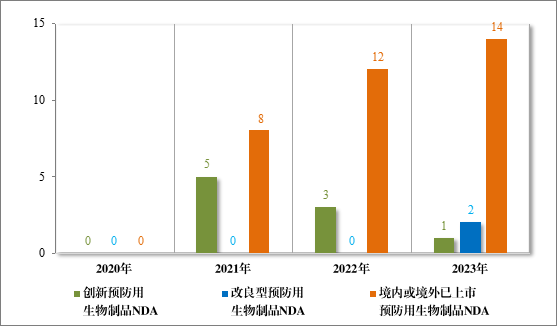
图 12 2020 年至 2023 年各注册分类预防用生物制品 NDA 受理情况(件)
治疗用生物制品IND 1070 件,包括创新治疗用生物制品 IND 833件(576 个品种),同比增加 32.85%;改良型治疗用生物制品 IND146件(75 个品种),同比增加 39.05%;境内或境外已上市治疗用生物制品 IND 91 件(46 个品种),同比增加 16.67%。
治疗用生物制品 NDA 178 件,包括创新治疗用生物制品 NDA 45件(29 个品种),同比增加 136.84%;改良型治疗用生物制品 NDA 38件(18 个品种),同比增加 137.50%;境内或境外已上市治疗用生物制品 NDA 95 件(58 个品种),同比增加 17.28%。
2023 年各注册分类治疗用生物制品 IND、NDA 受理情况详见表4,2020 年至 2023 年各注册分类治疗用生物制品 IND 受理情况详见图 14,2020 年至 2023 年各注册分类治疗用生物制品 NDA 受理情况详见图 15。
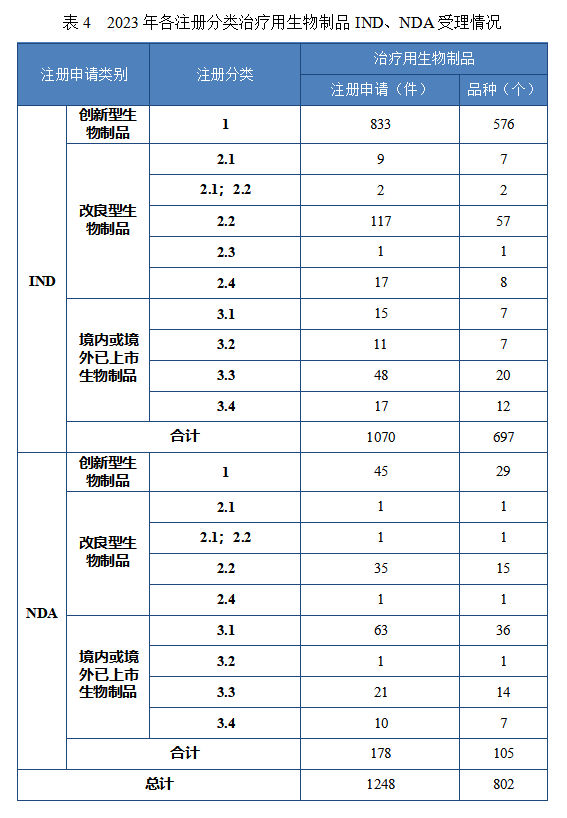
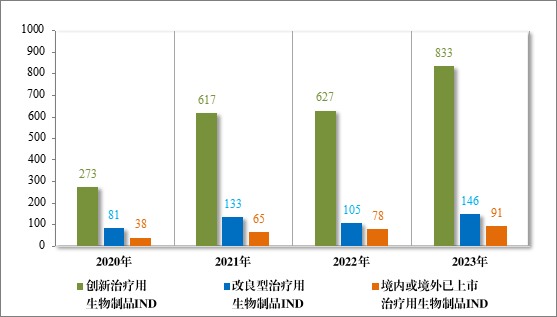
图 13 2020 年至 2023 年各注册分类治疗用生物制品IND 受理情况(件)
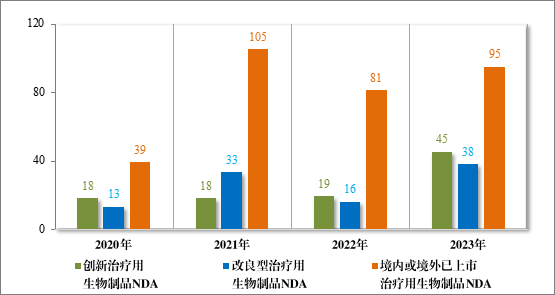
图 14 2020 年至 2023 年各注册分类治疗用生物制品 NDA 受理情况(件)
(三)直接行政审批类注册申请受理情况
2023 年受理直接行政审批类注册申请3745 件,同比增加22.11%;包括无需技术审评的补充申请 3367 件,同比增加 24.66%;临时进口注册申请 378 件,同比增加 3.28%。2019 年至 2023 年直接审批的各类别注册申请受理情况详见图 16。
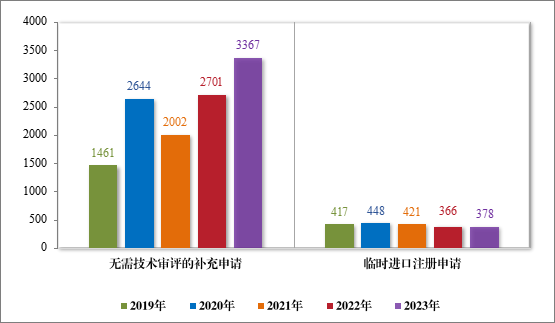
图 15 2019 年至 2023 年直接审批的各类别注册申请受理情况(件)
第二章 药品注册申请审评审批情况
(一)总体情况
2023 年,药审中心审结3注册申请共 15713 件(同比增加 28.80%),包括药品制剂注册申请 14523 件(同比增加 27.79%),化学原料药注册申请 1190 件(同比增加 42.51%)。14523 件药品制剂注册申请包含技术审评类注册申请 10642 件(同比增加 25.75%,包括 10633 件药品,9 件药械组合),直接行政审批类注册申请 3881 件(同比增加 33.74%)。2019 年至 2023 年注册申请审结量详见图 16。
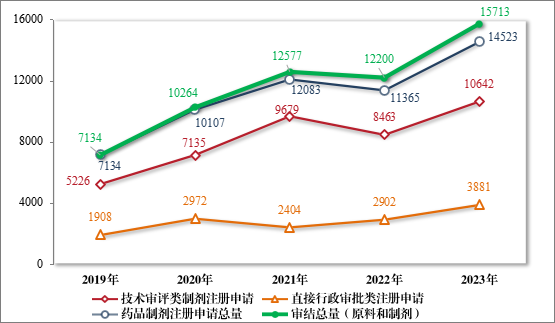
图 16 2019 年至 2023 年注册申请审结量(件)
截至 2023 年底,在审和待审的注册申请共 11059 件,其中包含原料药注册申请 2148 件;待申请人回复补充资料的注册申请共 2153件,其中包含原料药注册申请 622 件。
(二)技术审评类注册申请审结情况
3 本报告所称“审结”包括:完成技术审评报送国家局审批、完成技术审评后以国家局名义审批、不需要技术审评以国家局名义直接审批。“审结”不包含已完成技术审评后,因需申请人补充资料、发出补充资料通知书的注册申请(以下简称待申请人回复补充资料)。
2023 年审结的 10642 件技术审评类注册申请中,按药品类型计,中药注册申请 878 件,同比增加 131.05%;化学药品注册申请 7725件,同比增加 24.88%,占全部需技术审评审结量的 72.59%;生物制品注册申请 2030 件 ,同比增加 7.41%;药械组合注册申请 9 件。2019年至 2023 年需技术审评的各药品类型注册申请审结情况详见图 17。

图 17 2019 年至 2023 年需技术审评的各药品类型注册申请审结情况(件)
按审评序列计,2023 年审结 IND 2724 件,同比增加 18.43%;验证性临床试验申请 168 件,同比增加 22.63%;NDA 427 件,同比增加 26.71%;ANDA 2241 件,同比增加 60.76%;一致性评价申请 990件,同比增加 7.26%;补充申请 3541 件,同比增加 20.03%;境外生产药品再注册申请 538 件,同比增加 29.95%。2019 年至 2023 年需技术审评的各类别注册申请审结情况详见图 18。
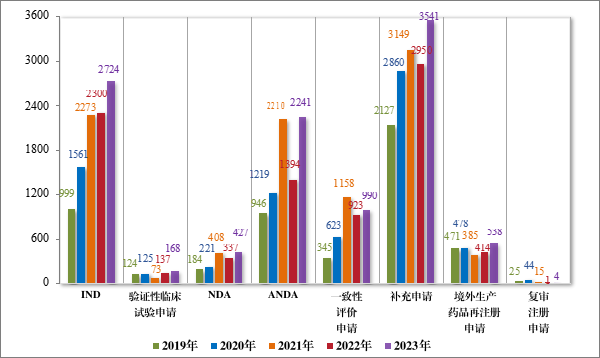
图 18 2019 年至 2023 需技术审评的各类别注册申请审结情况(件)
2023 年,药审中心采取多种措施提高审评效率,加快药品审评速度,以临床价值为导向,为患者提供更多的用药选择。
全年批准上市 1 类创新药 40 个品种(详见附件 1),其中 9 个品种(22.5%)通过优先审评审批程序批准上市,13 个品种(32.5%)为品附条件批准上市,8 个品种(20%)在临床研究阶段纳入了突破性治疗药物程序、4 个新冠治疗药物(10%)通过特别审批程序批准上市。
全年批准罕见病用药 45 个品种(未包括化药 4 类罕见病用药),其中 15 个品种(33.3%)通过优先审评审批程序得以加快上市(详见附件 2),1 个附条件批准上市。全年批准儿童用药产品 92 个品种,包含 72 个上市许可申请,其中 26 个品种(28%)通过优先审评审批程序得以加快上市(详见附件 3);另批准 20 个品种扩展儿童适应症,让更多儿童患者和千万家庭从中受益。
全年批准 CAR-T 细胞治疗产品 3 个,包括附条件批准伊基奥仑赛注射液、纳基奥仑赛注射液上市,附条件批准阿基仑赛注射液增加新适应症。
全年批准境外已上市、境内未上市的原研药品(化学药品 5.1 类、生物制品 3.1 类)86 个品种,其中 62 个为新批准上市,包括 1 个纳入临床急需境外新药名单内的品种,24 个为新增适应症,详见附件 4。
1. 中药注册申请审结情况
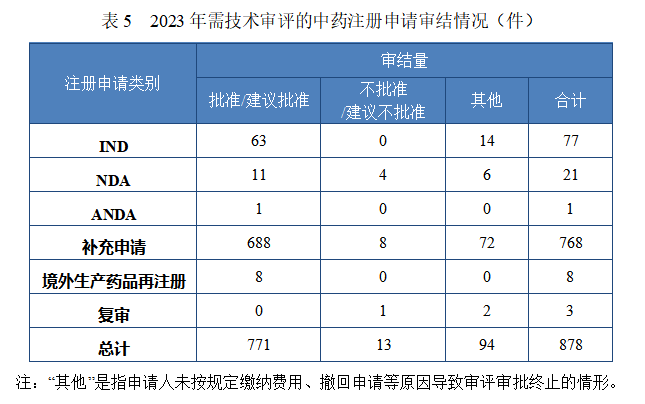
2023 年审结中药注册申请 878 件,同比增加 131.05%。按审评序列计,IND 77 件,同比增长 32.76%;NDA 21 件,同比增加 50.00%; ANDA 1 件。2023 年中药各类别注册申请审结情况详见表 5。
批准中药 IND 63 件,同比增长 40.00%,包括创新中药 IND 45件(39 个品种),同比增长 50.00%;改良型中药 IND 17 件(17 个品种),同比增长 30.77%;同名同方药 IND 1 件(1 个品种)。建议批准中药 NDA 11 件(10 个品种),同比增长 37.50%,包括创新中药 NDA 7 件(6 个品种);改良型中药 NDA 1 件(1 个品种);古代经典名方中药复方制剂 NDA 3 件(3 个品种)。建议批准中药 ANDA 1 件(1个品种),属中药同名同方药。
2023 年各注册分类中药IND、NDA 批准/建议批准情况详见表 6,2020 年至 2023 年各注册分类中药 IND 批准情况详见图 19,2020 年至 2023 年各注册分类中药 NDA、ANDA 建议批准情况详见图 20。
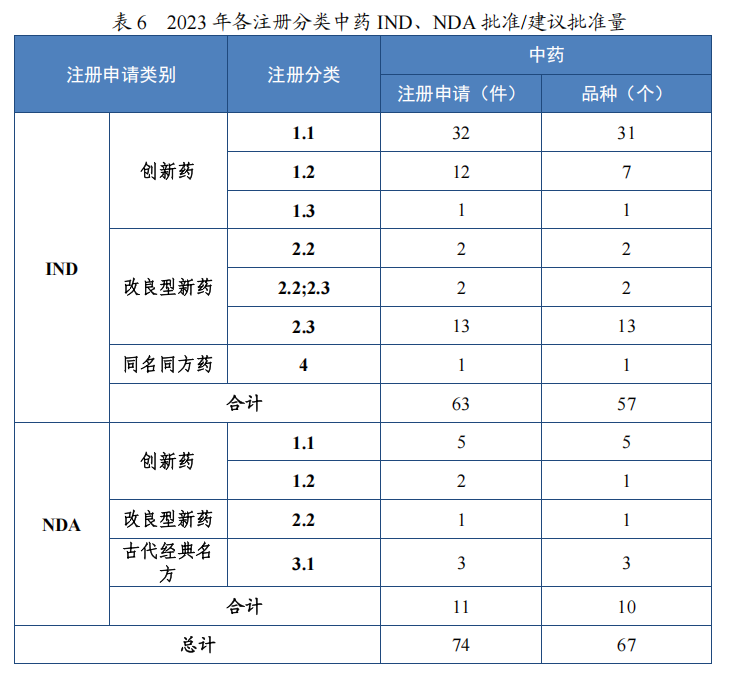
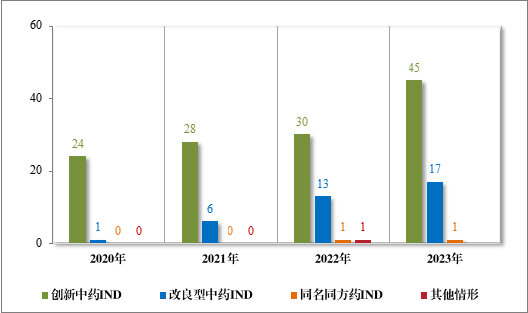
图 19 2020 年至 2023 年各注册分类中药IND 批准情况(件)

图 20 2020 年至 2023 年各注册分类中药NDA、ANDA 建议批准情况(件)
批准的 63 件中药 IND 中,涉及 13 个适应症领域,其中呼吸、消化药物较多,占中药 IND 批准量的 46.03%。2023 年批准中药 IND的适应症领域分布情况详见图 21。
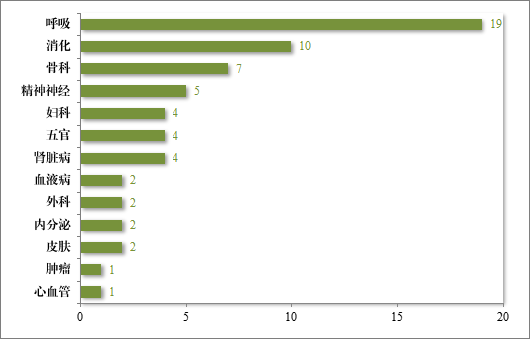
图 21 2023 年批准中药IND 的适应症领域分布情况(件)
建议批准的中药 NDA 11 件中,涉及 6 个适应症领域,其中消化药物较多,占中药 NDA 建议批准量的 36.36%。2023 年建议批准中药 NDA 的适应症领域分布情况详见图 22。
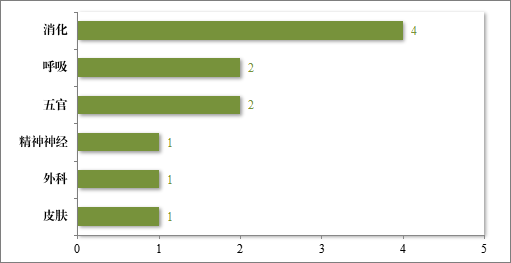
图 22 2023 年建议批准中药 NDA 的适应症领域分布情况(件)
2. 化学药品注册申请审结情况
2023 年审结化学药品注册申请 7725 件,同比增加 24.88%。按审评序列计,化学药品临床试验申请 1762 件,同比增加 16.92%;化学药品 NDA237 件,同比增加 21.54%;化学药品 ANDA 2240 件,同比增加 60.80%;化学药品一致性评价申请 990 件,同比增长 7.26%。 2023 年化学药品各类别注册申请审结情况详见表 7。
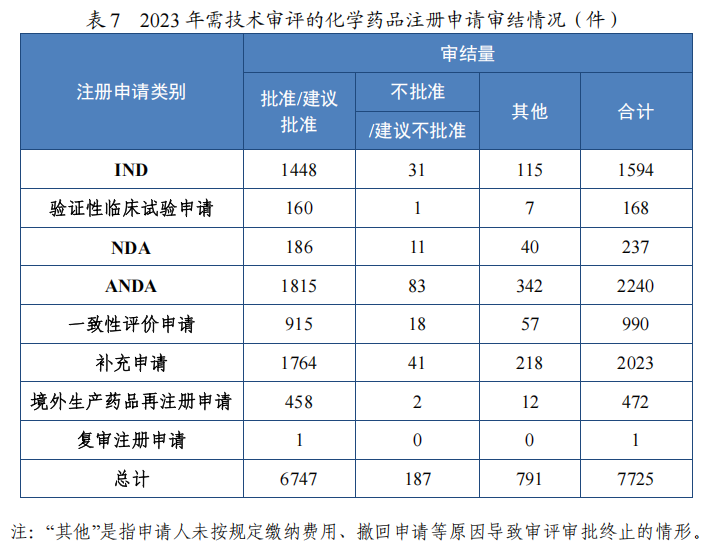
(1) 新药临床试验申请和新药上市申请
批准化学药品 IND 1448 件,同比增加 15.92%,包括创新化学药品 IND 1147 件(513 个品种),同比增加 13.12%;改良型化学药品 IND301 件(171 个品种),同比增加 27.54%。建议批准化学药品 NDA186 件(109 个品种),同比增加 21.57%,包括创新化学药品 NDA 38 件(20 个品种),同比增加 123.53%;改良型化学药品 NDA 39 件(25 个品种),同比减少 20.41%;5.1 类化学药品 NDA109 件(67 个品种),同比增加 31.33%。
2023 年各注册分类化学药品 IND、NDA 批准/建议批准情况详见表 8,2019 年至 2023 年各注册分类化学药品临床试验申请批准情况详见图 23,2019 年至 2023 年各注册分类化学药品 NDA 建议批准情况详见图 24。
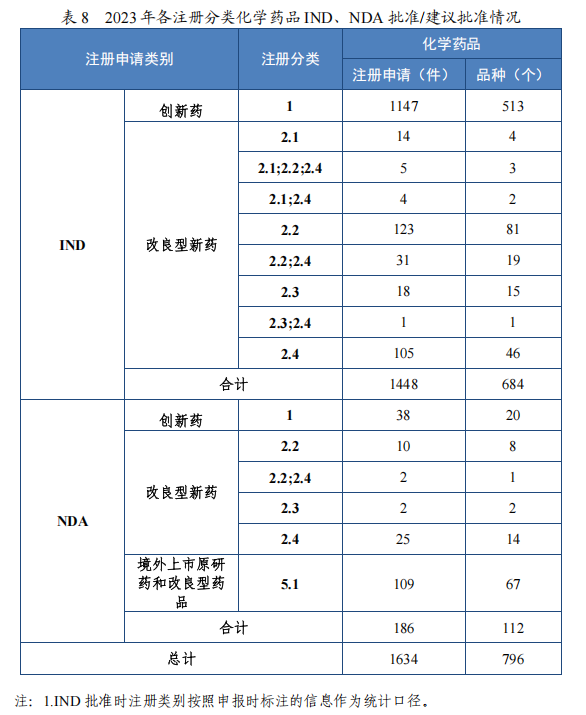

图 23 2019 年至 2023 年各注册分类化学药品 IND 批准情况(件)
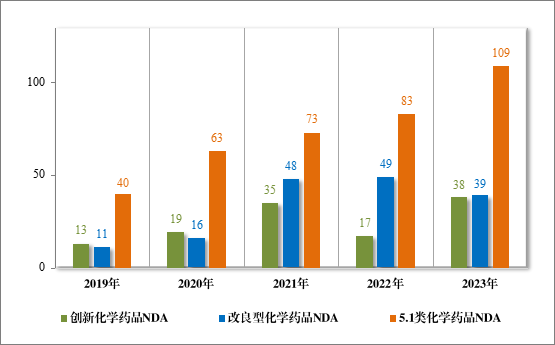
图 24 2019 年至 2023 年各注册分类化学药品 NDA 建议批准情况(件)
批准的化学药品 IND 1448 件中,抗肿瘤药物占 35.98%、皮肤及五官科药物占 12.29%,内分泌系统药物占 7.94%。2023 年批准化学药品 IND 的适应症领域分布情况详见图 25。

图 25 2023 年批准化学药品IND 的适应症领域分布情况(件)
建议批准的化学药品 NDA 186 件中,抗肿瘤药物占 22.04%、内分泌系统药物占 12.90%,消化系统疾病药物占 10.75%。。2023 年建议批准化学药品 NDA 的适应症领域分布情况详见图 26。
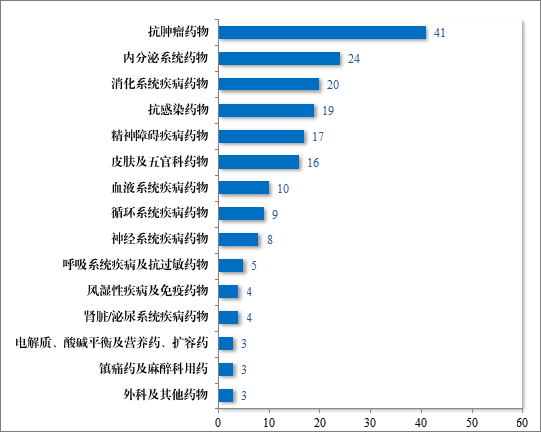
图 26 2023 年建议批准化学药品NDA 的适应症领域分布情况(件)
(2) 化学仿制药上市申请
2023 年建议批准化学仿制药上市申请1815 件,其中首仿品种246个,有效满足了临床患者对高质量仿制药的需求。
化学药品注册分类改革以来,药审中心按照与原研药品质量和疗效一致性的要求,累计建议批准化学仿制药 4545 件(864 个品种),涉及循环、抗感染、呼吸等 17 个治疗领域,具体见图 27。
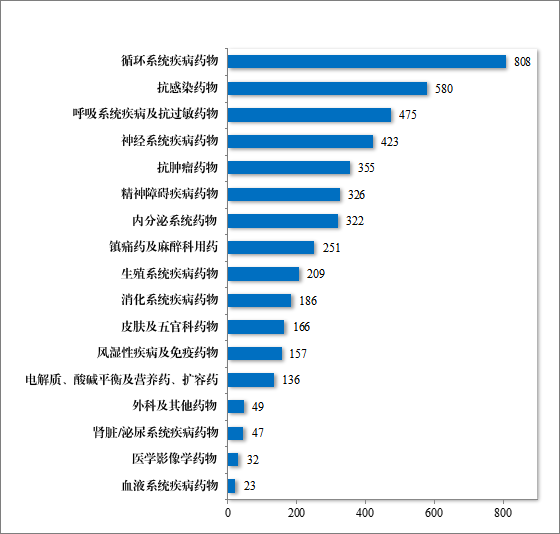
图 27 截至 2023 年批准化学药品新注册分类 ANDA 适应症领域分布情况(件)
2020 年,国家卫生健康委员会、国家发展改革委员会等十二部门发布了国家短缺药品清单,侧重在应对解决生产供应端短缺问题,保障药品供应。为落实短缺药品清单管理制度,做好短缺药品保供稳价工作,2023 年,药审中心共建议批准“国家短缺药品清单”药品 82件(18 个品种)。近五年累计批准上市的短缺药品共 109 件(25 个品种),见图 28,涉及生殖、循环、电解质、肿瘤等 9 个治疗领域,具体涉及适应症请见图 29,有效的保障了短缺药品的供应。
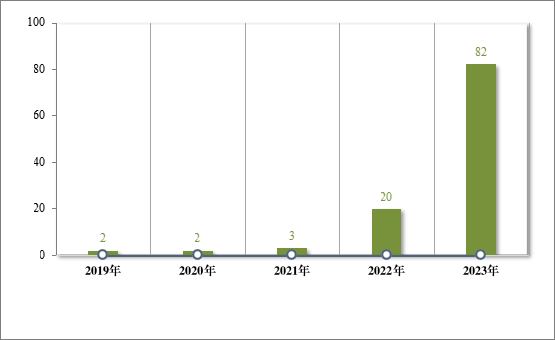
图 28 2019 年至 2023 年“短缺药品清单”药品批准情况
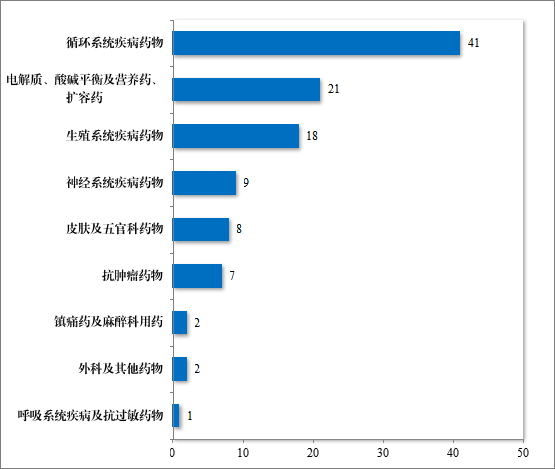
图 29 2023 年批准“短缺药品清单”药品适应症领域分布情况(件)
为进一步促进儿童适宜品种、剂型、规格的研发创制和申报审评,满足儿科临床用药需求,自 2016 年起,国家卫生健康委会同科技部、工业和信息化部、国家医保局和国家药监局研究制订了四批鼓励研发申报儿童药品清单,共有 136 个品种,包括口服溶液剂、口服混悬剂、颗粒剂等适宜儿童给药剂型。2023 年,药审中心建议批准属于鼓励研发申报儿童药品清单的儿童用仿制药 12 件(8 个品种)。2019 年以来累计建议批准属于鼓励研发申报儿童药品清单的儿童用仿制药 45件(18 个品种),见图 30,涉及神经、抗肿瘤、内分泌等 8 个治疗领域,具体涉及适应症情况请见图 31。
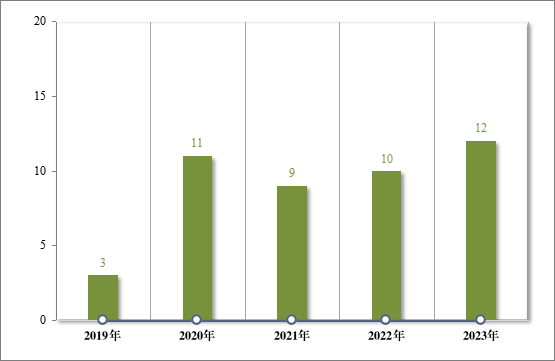
图 30 2019 年至 2023 年“鼓励研发申报儿童药品清单”药品批准情况(件)
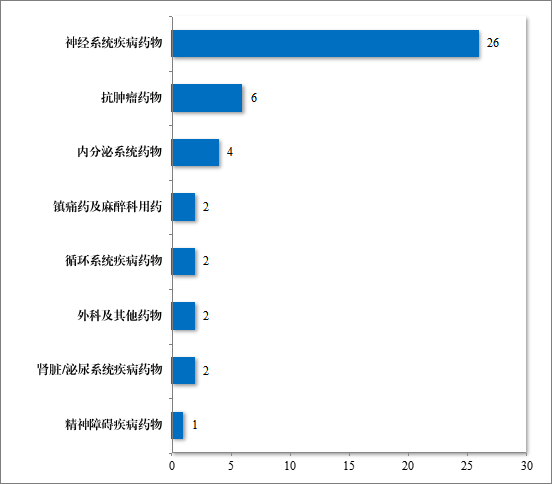
图 31 截至 2023 年批准“鼓励研发申报儿童药品清单”药品适应症领域分布情况(件)
(3) 化学仿制药质量和疗效一致性评价工作
2023 年,药审中心持续推进化学仿制药质量和疗效一致性评价工作,为患者提供高质量仿制药,共发布参比制剂目录 12 批,涉及804 个品规(497 个品种)。截至 2023 年 12 月 31 日,药审中心共收到参比制剂遴选申请信息 7955 条(3127 个品种)、自证信息 380 条(286 个品种),累计发布 74 批参比制剂目录,共 6714 个品规(2516个品种)。
2023 年审评通过一致性评价 915 件(326 个品种),其中,口服固体制剂通过 289 件(163 个品种);注射剂通过 626 件(163 个品种)。自化学仿制药质量和疗效一致性评价工作开展以来,累计通过一致性评价申请共 3797 件(共计 666 个品种),其中,口服固体制剂通过1836 件(计 417 个品种);注射剂通过 1961 件(计 249 个品种)。2019年至 2023 年一致性评价申请批准情况详见图 32。
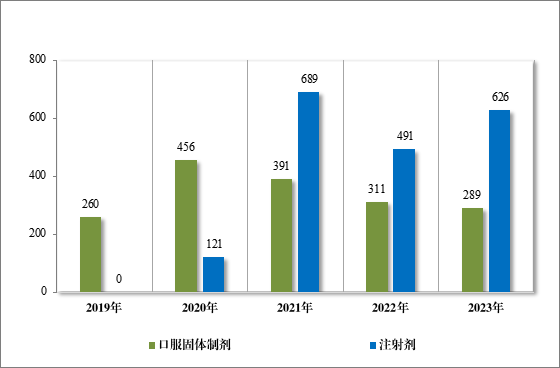
图 32 2019 年至 2023 年一致性评价申请批准量(件)
3. 生物制品注册申请审结情况
2023 年,审结生物制品注册申请 2030 件,同比增加 7.41%。其中,预防用生物制品 179 件和治疗用生物制品 1839 件,体外诊断试剂 12 件。
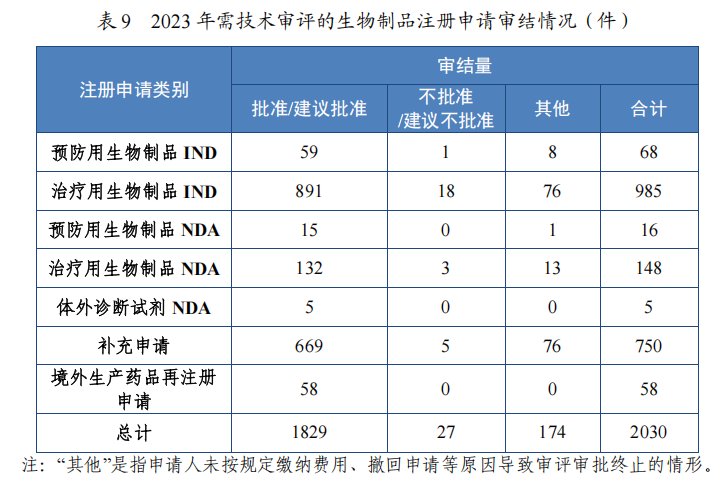
按审评序列计,IND 1053 件,同比增加 20.76%;NDA 169 件,同比增加 32.03%;补充申请 750 件;境外生产药品再注册申请 58 件。2023 年生物制品各类别注册申请审结情况详见表 9。
(1) 预防用生物制品
批准预防用生物制品 IND 59 件,包括创新预防用生物制品 IND 31 件(27 个品种),同比增加 72.22%;改良型预防用生物制品 IND 13件(10 个品种),同比增加 8.33%;境内或境外已上市预防用生物制品 IND 15 件(9 个品种),同比增加 50.00%。
建议批准预防用生物制品 NDA 15 件,包括创新预防用生物制品NDA2 件(2 个品种);境内或境外已上市预防用生物制品 NDA 11 件(9 个品种),另有 2 件(2 个品种)为原《药品注册管理办法》规定的已有国家药品标准的疫苗。
2023 年各注册分类预防用生物制品 IND、NDA 批准/建议批准情况详见表 10,2020 年至 2023 年各注册分类预防用生物制品 IND 批准情况详见图 33,2020 年至 2023 年各注册分类预防用生物制品 NDA建议批准情况详见图 34。

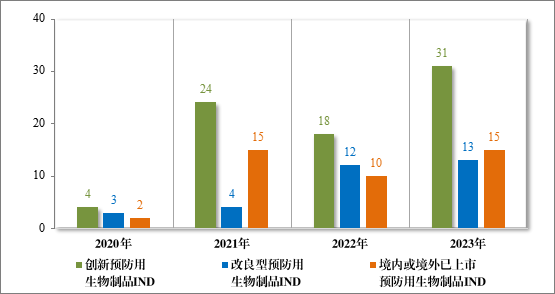
图 33 2020 年至 2023 年各注册分类预防用生物制品IND 批准情况(件)

图34 2020 年至2023 年各注册分类预防用生物制品NDA 建议批准情况(件)
(2) 治疗用生物制品
批准治疗用生物制品 IND 891 件,包括创新治疗用生物制品 IND 695 件(510 个品种),同比增加 25.68%;改良型治疗用生物制品 IND134 件(69 个品种),同比增加 20.72%;境内或境外已上市治疗用生物制品 IND 62 件(37 个品种),与去年持平。
建议批准治疗用生物制品 NDA 132 件,包括创新治疗用生物制品 NDA 19 件(15 个品种),同比增加 111.11%;改良型治疗用生物制品NDA 19 件(13 个品种);境内或境外已上市治疗用生物制品 NDA92 件(52 个品种),同比增加 84.00%,其中包括生物类似药(3.3 类)24 件(15 个品种)。(原 15 类 1 件,7 类 1 件)
2023 年各注册分类治疗用生物制品 IND、NDA 批准/建议批准情况详见表 11,2020 年至 2023 年各注册分类治疗用生物制品 IND 批准情况详见图 35,2020 年至 2023 年各注册分类治疗用生物制品 NDA建议批准情况详见图 36。
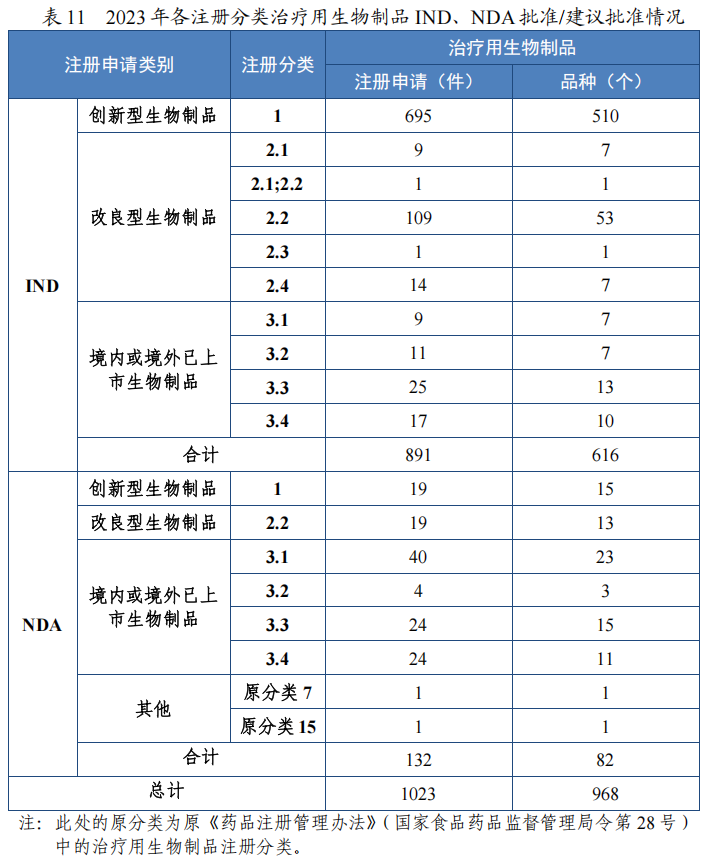

图 35 2020 年至 2023 年各注册分类治疗用生物制品IND 批准情况(件)
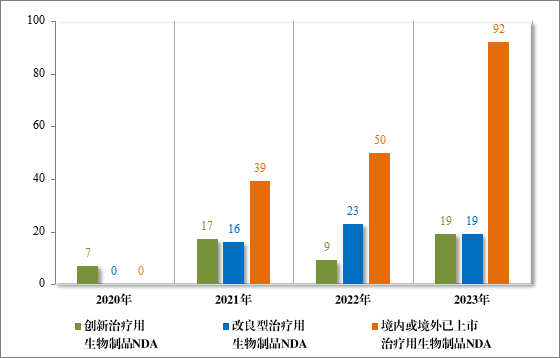
图 36 2020 年至 2023 年各注册分类治疗用生物制品NDA 建议批准情况(件)
批准的治疗用生物制品 IND891 件中,抗肿瘤药物占比 53.65%,皮肤及五官科药物占比 9.76%,血液系统疾病药物占比 6.06%。2023年批准治疗用生物制品 IND 的适应症领域分布情况详见图 37。
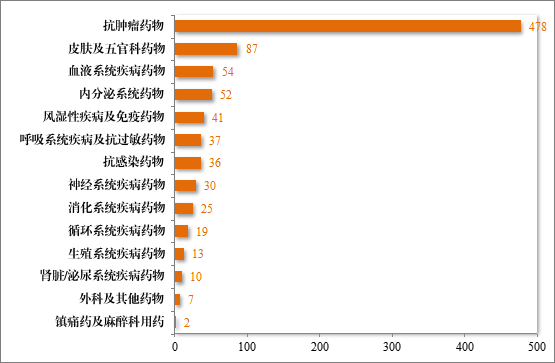
图 37 2023 年批准治疗用生物制品IND 的适应症领域分布情况(件)
建议批准的治疗用生物制品 NDA 132 件中,抗肿瘤药物占比32.58%,内分泌系统药物占比 19.70%,血液系统疾病药物 12.88%。2023 年建议批准治疗用生物制品 NDA 的适应症领域分布情况详见图38。
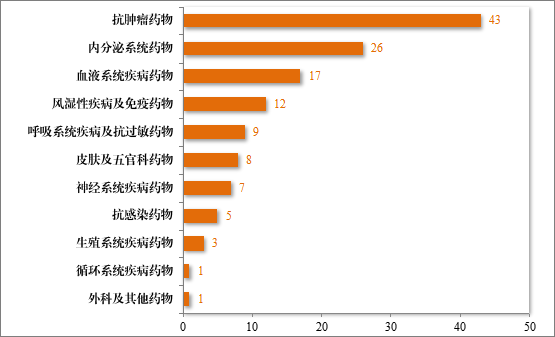
图 38 2023 年建议批准治疗用生物制品 NDA 的适应症领域分布情况(件)
(三)直接行政审批类注册申请审结情况
2023 年审结直接行政审批类注册申请3881 件,同比增加33.74%;其中无需技术审评的补充申请 3502 件,同比增加 37.60%;临时进口注册申请 379 件,同比增加 6.16%。2019 年至 2023 年直接审批的各类别注册申请审结情况详见图 39。
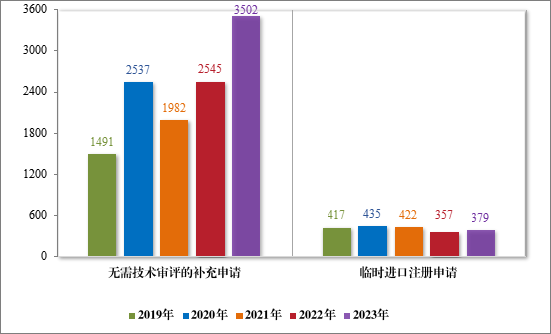
图 39 2019 年至 2023 年直接审批的各类别注册申请审结量(件)
第三章 加快新药好药上市,满足临床患者需求
2023 年度,药审中心通过药品加快上市注册程序,加强与申请人的沟通互动,缩短药物研发与技术审评时间,为患者提供更多治疗严重危及生命疾病、应对公共卫生事件的安全、有效、质量可控的临床用药。
(一)突破性治疗药物程序
药物临床试验期间,用于防治严重危及生命或者严重影响生存质 量的疾病且尚无有效防治手段或者与现有治疗手段相比有足够证据 表明具有明显临床优势的创新药或者改良型新药等,申请人可以在Ⅰ、 Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前申请适用突破性 治疗药物程序。对于适用突破性治疗程序的药物,其临床试验期间沟 通交流包括首次沟通交流、因重大安全性问题/重大技术问题而召开 的会议、药物临床试验关键阶段会议以及一般性技术问题咨询等,药 审中心优先配置资源进行沟通交流,加强指导并促进药物研发。
2023 年度,共收到突破性治疗药物程序申请 286 件,同意纳入突破性治疗药物程序 70 件(见附件 6),占申请数量的 24.5%,较 2022年增加 43%。排名前三的分别为抗肿瘤药物、神经系统疾病药物及消化系统疾病药物,具体适应症分布情况请见图 41。
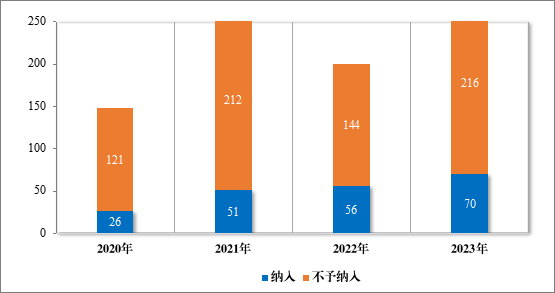
图 40 2020-2023 年突破性治疗药物程序纳入和不予纳入量(件)
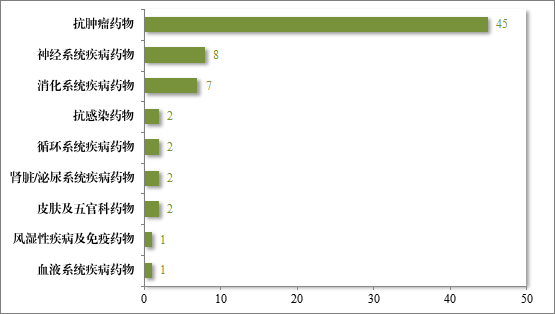
图 41 2023 年纳入突破性治疗药物程序适应症领域分布(件)
(二)附条件批准程序
药物临床试验期间,治疗严重危及生命且尚无有效治疗手段的疾病以及公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的,以及应对重大突发公共卫生事件急需的疫苗或 者国家卫生健康委员会认定急需的其他疫苗,经评估获益大于风险的,可基于替代终点、中间临床终点或早期临床试验数据而附条件批准上 市。附条件批准上市的目的是缩短药物临床试验的研发时间,使其尽 早应用于无法继续等待的危重疾病或公共卫生方面急需的患者。
2023 年共有 21 个药品附条件批准上市,其中 16 个药品为首次批准上市,5 个药品为新增适应症(见附件 7)。同时,在 2023 年,共有 10 个附条件批准上市药品完成了上市后研究,转为了常规批准。自 2020 年《药品注册管理办法》(总局第 27 号令)实施以来,共有95 个药品附条件批准上市,涉及 107 个适应症,包括抗肿瘤、抗新冠疫苗及治疗药物、血液系统疾病药物等,其中抗肿瘤药占比最多,为 79%,共有 19 个附条件批准上市药品完成了上市后研究,转为了常规批准,具体请见图 42、图 43。
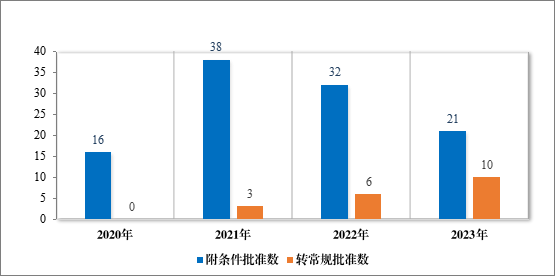
图 42 2020 年至 2023 年附条件批准和转为常规批准情况(按适应症计)

图 43 2020 年至 2023 年附条件批准药品适应症分布情况(按适应症计)
(三)优先审评审批程序
药品上市许可申请时,对于以下具有明显临床价值的药品,可以申请适用优先审评审批程序:(一)临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药;(二)符合儿童生理特征的儿童用药品新品种、剂型和规格;(三)疾病预防、控制急需的疫苗和创新疫苗;(四)纳入突破性治疗药物程序的药品;(五)符  附条件批准的药品;(六)国家药品监督管理局规定其他优先审评审批的情形。获得适用优先审评审批程序的上市注册申请的审评时限由常规程序的 200 日缩短为 130 日,其中临床急需的境外已上市境内未上市的罕见病药品审评时限为 70 日。
附条件批准的药品;(六)国家药品监督管理局规定其他优先审评审批的情形。获得适用优先审评审批程序的上市注册申请的审评时限由常规程序的 200 日缩短为 130 日,其中临床急需的境外已上市境内未上市的罕见病药品审评时限为 70 日。
2023 年度共纳入优先审评审批注册申请 108 件(80 个品种),同比增加 56.9%,具体纳入情况见图 44。2023 年有 85 件(59 个品种)注册申请按照优先审评审批程序批准上市。

图 44 2020 年至 2023 年根据现行《药品注册管理办法》纳入优先审评审批程序的注册申请情况(件)
自 2020 年《药品注册管理办法》(总局第 27 号令)实施以来,共有 372 个药品注册申请纳入优先审评审批程序,涉及的抗肿瘤药物、内分泌系统药物、皮肤及五官科药物等,其中抗肿瘤药占比最多,为 42%,具体请见图 45。
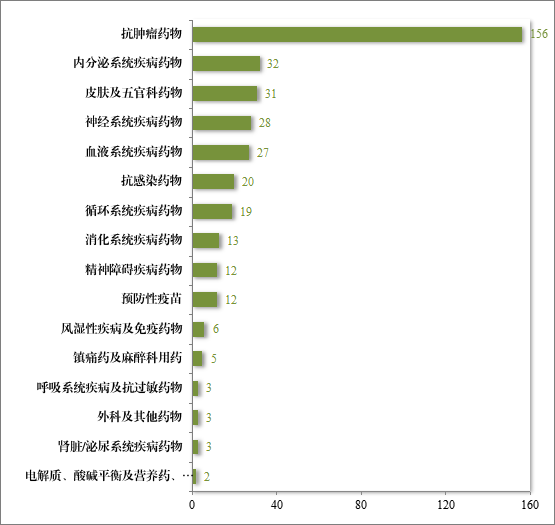
图 45 2020 年至 2023 年按现行《药品注册管理办法》纳入优先审评审批程序品种适应症分布情况
第四章 药品研发与审评沟通交流情况
沟通交流是在药物研发与注册申请过程中,申请人与药审中心审评团队之间针对所研发产品进行的不同形式的讨论,是药审中心服务于申请人的重要举措之一。
(一)沟通交流会议申请与办理情况
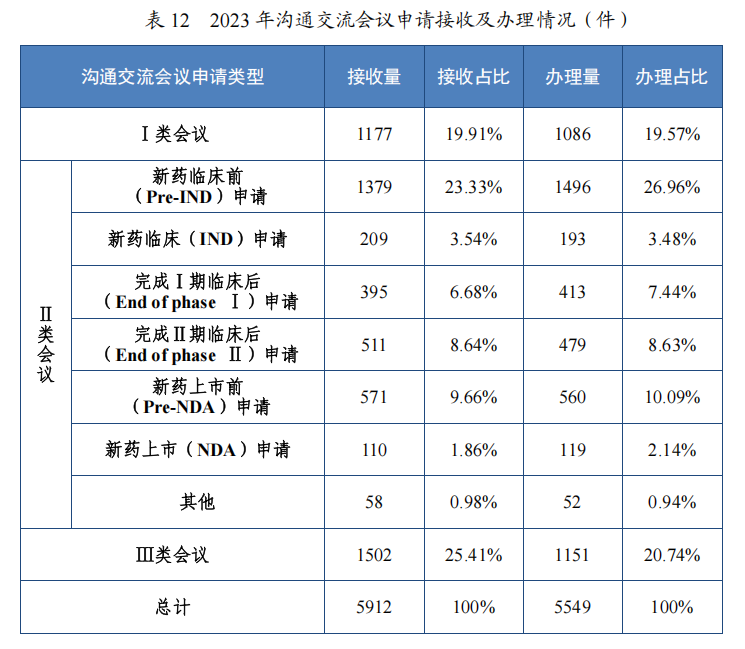
2023 年,药审中心共接收沟通交流会议申请 5912 件,同比增加20.06%,为 1607 家企业的 3710 个品种(按照申请人提交沟通交流申请时的药品名称计,下同)提供了沟通交流服务,办理沟通交流会议申请 5549 件,同比增加 27.59%,其中召开面对面会议/电话会议 612个,同比增加 24.29%。自 2017 年建立沟通交流会议制度以来,为 3180家企业的 11680 个品种在研发的关键阶段就重大问题进行了沟通指导,充分服务申请人,2019 年至 2023 年接收及办理沟通交流会议申请情况详见图 46。
2023 年,药审中心办理沟通交流会议申请 5549 件,召开面对面会议或电话会议 612 次,在药物研发关键阶段办理的Ⅱ类会议占比 54.69%,其中新药临床试验申请前(Pre-IND)申请占比 23.33%,新药上市许可申请前(Pre-NDA)申请占比 9.66%。2023 年沟通交流会议申请接收及办理情况详见表 12。

图 46 2019 年至 2023 年接收及办理沟通交流会议申请情况(件)
(二)一般性技术问题咨询情况
2023 年接收一般性技术问题咨询 16694 个,为 3831 家企业答疑一般性技术问题咨询 18173 个,经梳理总结,发布常见一般性技术问题及解答共 6 批 63 个。自 2017 年开展一般性技术问题咨询以来,为7033 家企业在研发过程和审评过程中就 108003 个一般性技术问题进行了答疑,总结发布 22 批 214 个共性问题4,同一企业累计提问数量分布情况详见表 13,2019 年至 2023 年接收及办理一般性技术问题咨询量详见图 47。
4 见链接:https://www.cde.org.cn/main/xxgk/listpage/07edef25f1e7354bfd8490baa0ce056b
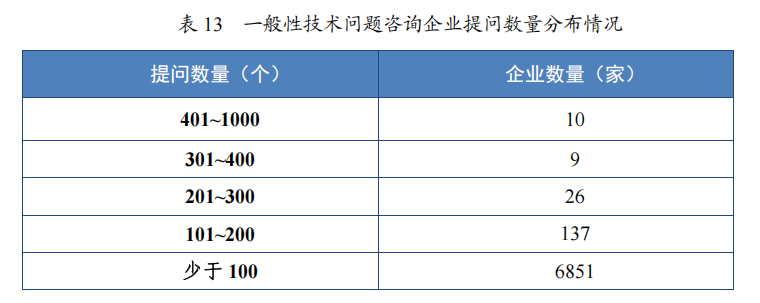
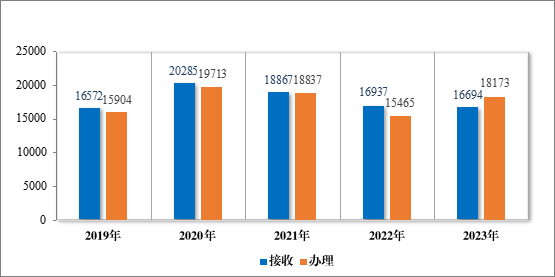
图 47 2019 年至 2023 年接收及办理一般性技术问题咨询量(个)
第五章 药品研发指导原则方面工作
(一)持续完善审评标准体系建设
2023 年药审中心制修订指导原则 74 个,新发布指导原则 60 个(详见附件 8),累计发布药品技术指导原则达 482 个5。
5 见链接:https://www.cde.org.cn/zdyz/index
1. 聚焦国际前沿技术领域,推动指导原则体系与国际先进技术标准深度融合
为推动 ICH Q13 指导原则在我国落地实施,发布了国内首个《化药口服固体制剂连续制造技术指导原则(试行)》。持续完善我国真实世界证据指导原则体系,继真实世界证据、真实世界数据的评价等技术要求,今年发布实施了《药物真实世界研究设计与方案框架指导原则(试行)》《真实世界证据支持药物注册申请的沟通交流指导原则(试行)》2 项技术指导原则。
加快完善放射性治疗药物评价体系,制定发布了《放射性体内治疗药物临床评价技术指导原则》《放射性治疗药物非临床研究技术指导原则(征求意见稿)》《放射性标记人体物质平衡研究技术指导原则(征求意见稿)》《放射性化学仿制药药学研究技术指导原则(征求意见稿)》等 4 项技术指导原则,推动相关产品加快研发上市。
加快完善细胞和基因治疗技术评价体系,制定发布了肿瘤主动免疫治疗产品、人源性干细胞、溶瘤病毒、基因治疗治疗血友病等 5 项技术指导原则。
2. 凝聚国际共识及监管实践,持续完善创新药物研发技术评价体系,有效缩短新药研发上市进程
首次将“以患者为中心”和基于“动物法则(Animal Rule)”药物注册理念纳入指导原则,标志着我国药物研发策略进入了新阶段。持续完善创新药物研发共性技术要求,发布实施了新药 III 期临床试验前药学沟通交流、化药复方药物临床试验设计、新药获益-风险评估、临床试验期间安全性信息评价、药物性肝损伤、单臂试验临床应用等 7 项技术指导原则。
3. 探索和丰富“三结合”注册审评证据体系,推动符合中医药特点的技术标准体系建设
围绕构建和完善“三结合”审评证据体系的工作任务,针对中药研发瓶颈和热点、难点问题,以问题为导向,不断创新工作方法,依托中药监管科学研究,广泛开展学术交流,充分发挥外部专家的作用,结 审评案例解剖麻雀,总结特点和规律,研究形成技术标准。2023年,发布《基于人用经验的中药复方制剂新药药学研究技术指导原则(试行)》等 5 项指导原则,同时,聚焦具有中医药治疗临床优势和特点的适应症,起草制定了糖尿病视网膜病变、紧张型头痛、小儿便秘等适应症临床研究技术指导原则,加快符
审评案例解剖麻雀,总结特点和规律,研究形成技术标准。2023年,发布《基于人用经验的中药复方制剂新药药学研究技术指导原则(试行)》等 5 项指导原则,同时,聚焦具有中医药治疗临床优势和特点的适应症,起草制定了糖尿病视网膜病变、紧张型头痛、小儿便秘等适应症临床研究技术指导原则,加快符 中药特点的疗效评价审评标准体系建设。
中药特点的疗效评价审评标准体系建设。
4. 加大力度解决公众及特殊群体的用药需求问题,提高常见疾病药物研发评价体系覆盖率
加快儿童用药研发进程,制定发布了《生理药代动力学模型在儿科人群药物研发中应用的技术指导原则》《成人用药数据外推至儿科人群的定量方法学指导原则(试行)》;为解决特殊群里用药吞咽困难,制定发布了《咀嚼片(化学药品)质量属性研究技术指导原则(试行)》;为加强说明书和标签规范管理,制定发布了《化学药品说明书及标签药学相关信息撰写指导原则(试行)》等 4 项技术指导原则。
首次推出《人乳头瘤病毒疫苗临床试验技术指导原则(试行)》,制定发布了首个《罕见疾病药物开发中疾病自然史》;在抗肿瘤药物领域制定发布了儿童抗肿瘤、晚期前列腺癌、卵巢癌 3 项技术指导原则;在血液系统疾病领域制定发布了慢性淋巴细胞白血病、急性髓系白血病 2 项技术指导原则;在内分泌系统、抗感染及抗病毒药物、消化系统疾病等领域制定发布了原发性胆汁性胆管炎、成人 2 型糖尿病、慢性乙型肝炎病毒感染、非阿片类术后镇痛药物等 6 项技术指导原则。
5. 总结仿制药申报共性问题,推动仿制药高质量发展
制定了国内外首个《化学药品仿制药溶液型滴眼剂药学研究技术指导原则》,针对产品安全性和质量可控性关键指标,制定发布了微生物限度、化学 成多肽、阿片类口服固体仿制药防滥用共 3 项技术指导原则。
成多肽、阿片类口服固体仿制药防滥用共 3 项技术指导原则。
(二)ICH 指南文件的转化实施
一是加快与国际药品注册技术要求接轨,逐步实现全球同步注册、同步研发。2017 年 6 月国家局加入 ICH 前,ICH 共发布了 57 个指导 原则,除《Q4B:药典》和《Q6B:质量标准:生物技术产品及生物 制品的检查方法及可接受标准》2 个 ICH 指导原则将在 2025 年版《中 国药典》中逐步实施外,其余 55 个ICH 指导原则均以发布适用公告、接受并翻译指导原则原文的形式实现充分实施。加入 ICH 之后至2023年 12 月,NMPA 积极选派专家参与了对旧版 ICH 指导原则的修订或增补的国际协调工作,共计 13 个,并参与制定新的 ICH 指导原则 11个,国家局均已原文采纳、充分实施。国家局已基本完成 ICH 当前全部 68 个指导原则的落地实施工作,这为中国患者可以及时分享全球药物创新的最新成果,用上放心的高质量药品奠定了基础。
二是全面参与 ICH 议题国际协调。截至 2023 年 12 月,ICH 正在活跃的协调议题共 32 个(详见附件 9),其中涉及有效性(Efficacy)指导原则 8 个,质量(Quality)指导原则 9 个,安全性(Safety)指导原则 2 个,多学科(Multidiscipline)指导原则 10 个,另有 3 个讨论组。国家局参与了每个 ICH 活跃议题的技术讨论和指导原则起草工作,并在国际协调过程中积极分享交流国内监管经验,了解借鉴其他监管机构监管良好实践。例如在参与 ICH M13 口服速释制剂生物等效性系列指导原则的协调过程中,结 中国丰富的仿制药审评和监管经验,国家局 ICH 专家踊跃发言,及时提供案例支持。
中国丰富的仿制药审评和监管经验,国家局 ICH 专家踊跃发言,及时提供案例支持。
三是紧跟国际学术前沿,及时引入转化 ICH 新理念、新方法、新工具、新标准。为适应制药产业和先进制造、纳米药物、人工智能等领域新兴技术的发展趋势,ICH 近年来协调的技术指南将对后续监管理念和监管方式带来调整。例如临床试验全球化、新的试验设计和新技术的应用推动了药物临床试验质量管理规范(GCP)的革新,ICH正在修订的 E6(R3)将提供更多的灵活性,提高患者参加临床试验的便利性;而 ICH 正在修订的《M4Q(R2):人用药品注册通用技术文档:质量》指导原则,将对药学注册申报资料的文件格式和组织框架进行重构,将为监管机构和业界带来一场变革。
第六章 积极推动监管科学研究,服务行业高质量发展
为鼓励医药产业创新,提高药品研发国际化水平,满足重大公共卫生应急状态下迫切的临床需求,推动中药传承创新,应对国际技术标准协调中新技术、新方法、新理念的挑战,提升自身监管能力,在国家局领导下,药审中心立足国际视野,聚焦国际监管科学前沿技术领域,大力推动药品监管科学研究相关工作,积极探索建立适合我国产业发展特点的监管新工具、新标准、新方法。
(一)药品监管科学的总体情况
2019 年 4 月,国家药监局启动实施中国药品监管科学行动计划,围绕建立科学、高效、权威的药品监管体系战略目标,深化药品审评审批制度改革创新,密切跟踪国际监管发展前沿,加快推进监管新工具、新标准、新方法研究和转化应用,先后启动了两批共 19 个监管科学重点项目,已转化药品监管相关新工具、新标准、新方法 187 项,为科学监管提供有力有效的技术支撑。其中,药审中心先后负责实施 8 个重点项目,参与实施 2 个重点项目,共计 21 个子课题,建立了66 项新工具、新方法、新标准,涉及基因和细胞治疗、纳米药物、放射性药物、真实世界数据、以患者为中心、连续制造、模型引导等,覆盖肿瘤、心脑血管、呼吸、自身免疫、重大慢病、传染病、儿童疾病和罕见病等疾病领域。
(二)药品监管科学项目的组织实施
药审中心立足药品审评工作实际,对标医药科学技术和国际药品监管科学前沿进展,提出监管科学重点研究领域的立项建议,经专家评审论证、国家药监局批准后正式立项。在组织实施阶段,药审中心以项目实施客观需求为依据,遴选出相关领域具有代表性、权威性的40 余家高校院所开展项目合作;以有效解决影响和制约药品创新、质量、效率的突出性问题为目标,确定研究内容和阶段性考核指标,建立项目动态跟踪管理机制,保证项目实施周期内,通过调研、论证、积累和创新,及时将成熟的监管经验转化为监管新工具、新标准、新方法,进一步增强审评工作的科学性、前瞻性和适应性,进一步提升审评的科学化、国际化和现代化水平,推动创新产品研发、注册与审评,更好满足公众健康需求。
(三)药品监管科学项目主要成效
1. 中药监管科学
(1) 在 2022 年发布的基于“三结合”注册审评证据体系下的沟通 交流指导原则基础上,优化中药注册程序,制定了关于加快古代经典 名方中药复方制剂沟通交流和申报的有关意见,一是加强研发关键节 点的沟通交流,将研发和申报划分为基准样品研究基本完成后、制备 工艺确定后/开展毒理研究前和申请上市许可前 3 个关键节点,建议 申请人在关键节点与药审中心进行沟通交流,并提出了相关资料要求;二是实行药学稳定性研究和毒理研究资料阶段性递交,加快技术审评。通过早期介入、研审联动等措施,加快相关品种的研发和申报进度。
(2) 发布了慢性胃炎、胃食管反流病、糖尿病视网膜病变、恶性肿瘤等临床研发评价指导原则,以具体适应症为突破口,发挥中医药理论和人用经验在中药新药研发中的指导作用。
(3) 应用真实世界证据探索有临床价值的药物和组方,以问题为导向,运用大数据和传统中医药融合,将独立、碎片化、局部的中医药信息整合,加强多来源中医药证据的快速收集、产生、评价、整合技术和方法学研究,探索具有临床价值的中药新药,计划制定真实世界在中药临床疗效评价中的应用技术指导原则。总结符合中药特色和优势的疗效评价指标特点以及中药特点的,以患者为中心的疗效评价指标的研制方法,为以患者为中心的中药临床疗效指标等新工具的设计和研发提供方法学的指导。计划起草以患者为中心的中药研发指导原则 。
(4) 构建“中药安全性(毒性)数据库”,已基本完成了毒性药材专题知识库的搭建,完成知识图谱发布与存储阶段;结合审评案例研究了以中医临床为导向的中药安全评价分类分级方法;形成法定药材标准中标示为有毒的药材名单(共计 152 种药材)
通过开展中药监管科学研究,目前已制定 11 项药品技术指导原则。加快构建“中药三结合”审评证据体系,加快确有临床价值的中药新药审评。
2. 化药监管科学
(1) 加快完善抗肿瘤药物研发指导原则体系,提出“以患者为中心、以临床价值为导向”的研发策略,助力抗肿瘤新药研发实现“突出重围、快人一步”。
(2) 集合国家纳米科学中心、北京大学医学部、中检院等科研和监管领域专家力量,建立了涵盖聚合物纳米粒子、胶束、脂质体、树枝状大分子、金属纳米粒子、固体脂质纳米粒子等的纳米类药物的质量控制研究、临床前研究、临床安全性和有效性的评价体系,制定了 5 项纳米药物、脂质体类纳米药物等药学和非临床评价技术指导原则,促进国内利培酮微球、紫杉醇胶素、米托蒽醌脂质体等纳米药物的研发和广泛应用,产生巨大的临床价值和经济效益。
(3) 加快发布适用于儿童用药的指导原则,促进和保障一大批临床急需儿童用药上市,儿童适宜剂型少、规格少等问题也进一步得到缓解,2019 年以来,儿童用药申报量、获批量均明显上升,儿童用药可及性大大提高。
(4) 鼓励以临床价值为导向的放射性药品创新,成立放射性药品专项工作组,印发改革完善放射性药品审评管理工作方案,制定不同放射性元素的化学仿制放射药个药技术审评要点和指导原则,初步构建放射性药品审评标准体系。
3. 生物制品监管科学
(1) 建立完善细胞和基因治疗研发与评价技术标准体系,制定 16 项技术指导原则,助力我国批准 4 款CAR-T 药物,批准多个 CAR-T和 CAR-NKT 产品开展临床试验,治疗的适应症进一步拓展,靶点分布进一步丰富。
(2) 起草了新型冠状病毒变异株预防用疫苗研发与评价指导原则、新型冠状病毒预防用疫苗临床研究及评价的考虑要点技术指导原则等,进一步完善新冠预防药物研发和评价指导原则体系,在保证我国新冠疫苗研发监管要求以及标准发布速度与国际保持一致基础上,科学审评同步优化升级流程制度,促进了研发评价体系与时俱进,出色地完成了新冠疫苗的应急审评工作。
(3) 积极加强国内外学科交流,通过中国生物制品质量控制大会,围绕细胞和基因治疗产品、疫苗及血液制品、治疗用重组生物制 品的前沿基础研究、生物技术转化创新、生物制品生命周期审评和上 市后监管等,与多个国家和地区的生物制品监管机构与学界开展交流,分享我国的监管经验。
4. 交叉学科领域监管科学
(1) 真实世界研究在药物临床研发中的合理应用及其方案设计 是使用真实世界数据及证据支持药物监管决策的关键问题之一。2020 年,中心制定并发布全球首个真实世界证据支持药物研发与审评指导 原则,2021 年发布用于产生真实世界证据的真实世界数据指导原则, 为进一步补充细化技术要求,指导申办者科学理设计真实世界研究,明确真实世界研究方案撰写技术要求,本年度发布了药物真实世界研 究设计与方案框架指导原则,为后续 ICH M14 协调提供技术储备。 与此同时,也发布了真实世界证据支持药物注册申请的沟通交流指导 原则,阐述使用真实世界证据在关键时间节点开展沟通交流的核心问题,帮助申请人提高研发效率,促进在真实世界研究领域的深入实践。
(2) 发布化药口服固体制剂连续制造技术指导原则。项目实施期间开展的连续制造相关的交流、研讨和培训等系列工作,有助于企业及监管机构对连续制造的技术和监管要求有更深入的认识和理解,及早发现 ICHQ13 落地实施存在的困难及可能存在的问题,形成实施建议,极大推动了 ICHQ13 国内的落地实施。助力 2 款采用连续生产技术产品获批上市,4 款获批临床。
(3) 以鼓励创新为导向,开展以患者为中心的临床试验设计、实施、风险获益评价的研究,制定以患者为中心的药物开发指导原则 3 项;首次在国内明确患者体验数据(patient experience data, PED)的定义和分类,首次构建了应用患者体验数据的获益-风险评估的理论框架,实践“以患者为中心”的理念,了解特应性皮炎/湿疹对患者的影响与治疗现状,制定全球首个特应性皮炎治疗药物临床试验技术指南,有助于在特应性皮炎/湿疹药物的临床开发、监管决策中充分纳入患者体验数据。
探索启动“以患者为核心的罕见疾病药物研发”试点工作,以罕见疾病为抓手,落实“以患者为核心的药物研发”理念,在药物研发的全程,引入罕见疾病患者的观点,倾听患者声音,切实调动罕见疾病患者在药物研发过程中的参与意识,发挥患者观点对药物研发的指导作用,在研发全程申请按程序要求开展相应的沟通交流,按照加快上市注册程序要求缩短审评审批周期,促进相应罕见病药物快速上市。
第七章 药品研发与技术审评宣贯与培训
2023 年,药审中心梳理企业关心的药品审评政策法规、技术指南和流程管理咨询热点问题,分专题为申请人集中讲解资料撰写及受理关注点、审评考虑、发补共性问题等,同时穿插宣贯以患者为中心、儿童用药等相关新发布的技术指导原则,以线上线下相结方式开展对外培训专题 24 场,发布视频回放 12 期,初步形成了“线上直播+视频回放”培训方式,让申请人更好了解中心审评流程、审评要求和审评依据,让审评工作更加公开、透明,具体清单详见附件 10。
线上培训重点开展了审评业务流程及技术要求讲解 12 场,辐射受众超过 11 万人。在审评业务流程方面系统讲解了沟通交流、注册受理与电子申报、核查检验、常规审评流程、加快审评流程以及审评过程中的书面发补等药品审评业务专题,为申请人讲理念、讲要求、讲问题、讲实操,覆盖药品注册业务全流程;宣贯创新药研发、中药传承创新、临床急需药物研发等相关政策法规、改革成果、技术指导原则等,积极对外传递中心鼓励创新的新举措、新办法,激发我国医药产业研发动力,促进了新药研发和注册申报。
线下培训聚焦服务国家区域发展战略,聚焦业界关注的问题,精心设计培训课程并选派业务骨干进行授课,开展线下培训为主培训 12 场,线下参会人员近 8000 人。赴京津冀地区开展了 ICH《E6(R3):药物临床试验质量管理规范》、抗肿瘤创新药培训班;分别赴长三角、大湾区开展“细胞和基因治疗产品临床研发技术指导原则主题”、“化学新药药学沟通交流培训会”、“《中药注册管理专门规定》培训班”、“生物制品变更管理技术指导培训会”;分别赴辽宁、吉林、黑龙江开展“支持东北医药产业发展药品注册技术系列培训班”, 联合辽宁、吉林和黑龙江三个省局共同主办了化药、生物制品、中药三个专题,严格按照“提前介入,一企一策,全程指导,研审联动”的要求,创新性将药品注册技术培训与企业座谈会有机结合,对东北地区重点品种和重点医药企业提供有针对的培训和指导,助力振兴东北生物医药产业创新和高质量发展需要。
第八章 2023年度药品审评主要工作回顾
(一) 自 2023 年 1 月 1 日起,根据国家药监局发布的《关于实施药品注册申请电子申报的公告》(2022 年第 110 号),申请人提交国家药监局审评审批的药品注册申请以及审评过程中补充资料等,调整为以电子形式提交申报资料。
(二) 2023 年 1 月 29 日,国家药监局发布关于适用《Q3D(R2):元素杂质》《M10:生物分析方法验证及样品分析》国际人用药品注册技术协调会指导原则的公告(2023 年第 16 号),规定自 2023 年 7月 29 日起开展的相关研究(以实验记录时间点为准)适用 Q3D(R2)指导原则,自 2023 年 7 月 29 日起开展的相关研究(生物样品分析原始记录时间点为准)适用 M10 指导原则。
(三) 2023 年 2 月 10 日,国家药监局发布《中药注册管理专门规定》(2023 年第 20 号),该规定充分吸纳药品审评审批制度改革成熟经验,结合疫情防控中药成果转化实践探索,借鉴国内外药品监管科学研究成果,全方位、系统地构建了中药注册管理体系。
(四) 2023 年 3 月 22 日,国家药监局发布关于适用《S1B(R1):药物致癌性试验》和《E14-S7B 问答:致 QT/QTc 间期延长及潜在致心律失常作用的临床与非临床评价问答》国际人用药品注册技术协调会指导原则的公告(2023 年第 33 号),规定自 2023 年 3 月 22 日起开始的相关研究,均适用 S1B(R1)指导原则,自 2023 年 7 月 31日起,启动的药物临床研究相关要求适用 E14-S7B 问答指导原则。
(五) 2023 年 3 月 24 日,国家药监局发布《关于发布化学仿制药参比制剂调整程序的公告》(2023 年第 35 号),进一步完善了仿制药参比制剂管理。
(六) 2023 年 3 月 31 日,药审中心发布《药审中心加快创新药上市许可申请审评工作规范(试行)》,该规范依托于现有工作程序,集中审评资源靠前服务指导,鼓励儿童专用创新药、用于治疗罕见病的创新药以及纳入突破性治疗药物程序的创新药研发,加快创新药上市速度。
(七) 2023 年 4 月 12 日,药审中心发布儿童用药技术审评临床外聘专家名单,扩大儿童用药专业领域专家力量。
(八) 2023 年 4 月 18 日,药审中心发布《儿童用药沟通交流中 I类会议申请及管理工作细则(试行)》,规定对儿童临床试验计划或儿童临床试验结果的沟通交流会议申请,可按照 I 类会议开展沟通交流。
(九) 2023 年 4 月 24 日,国家药监局发布关于适用《E19:在特定的上市前后期或上市后临床试验中选择性收集安全性数据》国际人用药品注册技术协调会指导原则的公告(2023 年第 56 号),规定自 2023 年 10 月 21 日起,启动的药物临床试验相关要求适用 E19。
(十) 2023 年 4 月 25 日,国家局发布《国家药监局关于改革完善放射性药品审评审批管理体系的意见》(国药监药注〔2023〕20 号),全面启动放射性药品审评审批改革工作,药审中心建立专项工作组,增选放射性药品外聘专家,立项起草放射性药品研发指导原则。
(十一) 2023 年 5 月,为积极支持国家重大战略,服务区域医药产业创新发展,药审中心按照“整合职能、强化服务、融合发展、相互支撑”的原则,联合两个分中心共同制定了“药物创新研发重点项目工作方案”,建立了“药品审评会商机制”,针对区域研发重点项目,创新工作机制,主动靠前服务,积极支持具有重大临床价值、处于科技前沿、自主原始创新的创新药的研发和转化
(十二) 2023 年 5 月 31 日,国家药监局发布《已上市药品说明书增加儿童用药信息工作程序(试行)》(2023 年第 68 号),该程序旨在完善已上市药品说明书儿童用药信息,提升儿童安全用药水平。
(十三) 2023 年 7 月 3 日,药审中心发布《化学原料药受理审查指南(试行)》(2023 年第 38 号),进一步规范化学原料药申报与受理审查工作。
(十四) 2023 年 7 月 4 日,国家药监局发布关于适用《M10:生物分析方法验证及样品分析》国际人用药品注册技术协调会指导原则问答文件和常见问题解答文件的公告(2023 年第 84 号),规定自 2023年 7 月 29 日起开始的相关研究(以生物样品分析原始记录时间点为准),均适用 M10 问答文件和常见问答解答文件。
(十五) 2023 年 7 月 5 日,国家药监局发布《药品标准管理办法》(2023 年第 86 号),该规范旨在加强药品标准管理,建立最严谨的药品标准,保障药品安全、有效和质量可控,促进药品高质量发展。
(十六) 2023 年 8 月,药审中心平稳有序、安全高效完成从国贸到北京经开区的办公地址搬迁工作。整体搬迁过程平稳有序、安全高效,仅暂停对外办公 5 日,并提前 1 日恢复“申请人之窗”运行,最大程度降低了搬迁对审评工作的影响。
(十七) 2023 年 8 月 25 日,国家药监局发布关于适用《Q12:药品生命周期管理的技术和监管考虑》国际人用药品注册技术协调会指导原则的公告(2023 年第 108 号),Q12 为药品上市后变更管理提供了新的实现方法和监管工具,申请人可以按照目前我国变更管理的相关法规规章和指导原则进行变更管理,也可以在提交上市申请和/或补充申请时采用 Q12 提供的新方法进行变更管理。
(十八) 2023 年 9 月,药审中心起草中药和化学药品生物制品《沟通交流申请资料要求》并对外征求意见,建立沟通交流申请人自评估机制,完善内部工作规范,多次组织召开沟通交流企业座谈会广泛听取意见建议,不断优化完善沟通交流机制。
(十九) 2023 年 9 月 5 日,国家药监局发布关于适用《Q9(R1):质量风险管理》国际人用药品注册技术协调会指导原则的公告(2023年第 114 号),规定自 2024 年 3 月 4 日后,上市许可持有人开展的质量风险管理活动,均适用《Q9(R1):质量风险管理》指导原则。
(二十) 2023 年 9 月 5 日,国家药监局发布关于适用《S12:基因治疗产品非临床生物分布的考虑》国际人用药品注册技术协调会指导原则的公告(2023 年第 115 号),规定自本公告发布之日起开始的非临床研究适用 S12 指导原则。
(二十一) 2023 年 9 月 15 日,国家药监局批准氟[18F]贝他苯注射液上市,该品种是我国首个用于阿尔兹海默病患者早期、精准、无创诊断的 Aβ-PET 显像剂,是中国近二十年来首个获批的放射性药物仿制药。
(二十二) 2023 年 9 月 20 日,药审中心发布《微型片剂(化学药品)药学研究技术指导原则(征求意见稿)》,该指导原则为全球药品监管机构首发,旨在支持儿童用药品研发创新。
(二十三) 2023 年 10 月 12 日,药审中心发布《药物临床试验方案提交与审评工作规范》,该规范旨在提高药品注册申请人撰写临床试验方案的质量,规范临床试验方案有关沟通交流和各类注册申请,提高临床试验方案的审评质量。
(二十四) 2023 年 10 月 13 日,国家药监局发布《国家药监局关于无参比制剂品种仿制研究的公告》(2023 年第 130 号)及政策解读,药审中心同步发布《无参比制剂品种开展仿制研究的技术要求和申报资料要求(试行)》、《无参比制剂品种开展仿制研究的沟通交流申请资料要求(试行)》,为无参比制剂品种仿制研究提供了申报和研究路径。
(二十五) 2023 年 10 月 13 日,国家药监局发布《关于化学原料药再注册管理等有关事项的公告》(2023 年第 129 号),明确了化学原料药批准通知书发放、化学原料药再注册、注销化学原料药批准证明文件的相关程序。
(二十六) 2023 年 10 月 31 日,国家药监局发布《药品说明书适老化及无障碍改革试点工作方案》(2023 年第 142 号),决定在部分口服、外用等药品制剂中开展说明书及无障碍改革试点。11 月 24 日,药审中心配套发布《药品说明书(简化版)及药品说明书(大字版)编写指南》和《电子药品说明书(完整版)格式要求》(2023 年第 56号),12 月 26 日,国家药监局公布第一批药品说明书适老化及无障碍改革试点名单,涉及 657 个品种。
(二十七) 2023 年 11 月,为了充分发挥专家在审评决策中的重要作用,解决专家资源与审评量不匹配的问题,药审中心明确了外聘专家动态管理、及时补充的工作机制,为进一步补充专家力量,充分发挥专家在技术审评中的作用奠定了基础。
(二十八) 2023 年 11 月 16 日,药审中心发布《自体 CAR-T 细胞治疗产品药学变更研究的问题与解答》,旨在更好地引导 CAR-T 类细胞治疗产品药学变更的研究与申报
(二十九) 2023 年 11 月 22 日,药审中心发布了《关于加快古代经典名方中药复方制剂沟通交流和申报的有关措施》,对按古代经典名方目录管理的中药复方制剂(中药 3.1 类)采取早期介入、研审联动等措施,加快相关品种的研发和申报进度。
(三十) 自 2023 年 12 月 1 日起,药审中心分别在两个分中心设立了对外受理服务窗口,并协助药审中心开展区域药品注册申请的受理工作,截至年底,长三角分中心与大湾区分中心分别受理区域内 136、71 件注册申请。
(三十一) 2023 年 12 月 14 日,国家药监局发布关于适用《Q13:原料药和制剂的连续制造》国际人用药品注册技术协调会指导原则的公告(2023 年第 158 号),规定自 2024 年 6 月 13 日开始的相关研究(以试验记录时间点为准),均适用 Q13 指导原则。
结语
路虽远,行则将至;事虽难,做则必成。新时代使命光荣,新征程任务艰巨。2024 年,药审中心将按照党的二十大精神,根据《“十四五”国家药品安全及促进高质量发展规划》,围绕国家局相关重点工作部署,深入推进全面从严治党,以高质量党建服务保障高质量发展,以防范利益冲突为抓手,扎实推进党风廉政建设,深化药品审评审批制度改革,全面提高科学审评质量和效率,加强现代化审评体系建设,深入推进审评标准与国际接轨,全力推动中药审评审批机制改革,促进中药传承创新发展,科学高效开展化学仿制药上市、一致性评价以及补充申请相关审评工作,服务国家发展战略,促进生物医药产业创新发展,聚焦前沿技术领域,全面加强药品监管科学研究,加强信息化建设,以智慧监管助力审评现代化,加强人才队伍建设,奋力推进药品监管各项工作,以更加优异的成绩,为保护和促进公众健康做出更大的贡献。
附件8 2023年药审中心发布的指导原则
2023 年药审中心发布的指导原则
| 序号 | 通告号 | 指导原则名称 | 发布时间 |
|---|---|---|---|
| 1 | 2023 年第 1 号 | 《慢性淋巴细胞白血病新药临床研发技术指导原则》 | 2023/1/19 |
| 2 | 2023 年第 2 号 | 《溶瘤病毒产品药学研究与评价技术指导原则(试行)》 | 2023/2/13 |
| 3 | 2023 年第 3 号 | 《急性髓系白血病新药临床研发技术指导原则》 | 2023/2/13 |
| 4 | 2023 年第 4 号 | 《原发性胆汁性胆管炎治疗药物临床试验技术指导原则》 | 2023/2/13 |
| 5 | 2023 年第 5 号 | 《药物真实世界研究设计与方案框架指导原则(试行)》 | 2023/2/16 |
| 6 | 2023 年第 6 号 | 《真实世界证据支持药物注册申请的沟通交流指导原则(试行)》 | 2023/2/16 |
| 7 | 2023 年第 7 号 | 《咀嚼片(化学药品)质量属性研究技术指导原则(试行)》 | 2023/2/14 |
| 8 | 2023 年第 8 号 | 《化学药品仿制药溶液型滴眼剂药学研究技术指导原则》 | 2023/2/16 |
| 9 | 2023 年第 9 号 | 《放射性体内治疗药物临床评价技术指导原则》 | 2023/2/15 |
| 10 | 2023 年第 10 号 | 《成人 2 型糖尿病药物临床研发技术指导原则》 | 2023/2/21 |
| 11 | 2023 年第 11 号 | 《非无菌化学药品及原辅料微生物限度研究技术指导原则(试行)》 | 2023/2/21 |
| 12 | 2023 年第 12 号 | 《化学成多肽药物药学研究技术指导原则(试行)》 | 2023/2/21 |
| 13 | 2023 年第 13 号 | 《单臂临床试验用于支持抗肿瘤药上市申请的适用性技术指导原则》 | 2023/3/14 |
| 14 | 2023 年第 14 号 | 《晚期前列腺癌临床试验终点技术指导原则》 | 2023/3/14 |
| 15 | 2023 年第 15 号 | 《化药复方药物临床试验技术指导原则》 | 2023/3/17 |
| 16 | 2023 年第 16 号 | 《药物临床试验期间安全性信息汇总分析和报告指导原则(试行)》 | 2023/3/17 |
| 17 | 2023 年第 17 号 | 《药物临床试验期间安全性数据快速报告常见问答(2.0 版)》 | 2023/3/17 |
| 18 | 2023 年第 18 号 | 《阿片类口服固体仿制药防滥用药学研究技术指导原则(试行)》 | 2023/3/17 |
| 19 | 2023 年第 19 号 | 《化药口服固体制剂连续制造技术指导原则(试行)》 | 2023/3/21 |
| 20 | 2023 年第 20 号 | 《化学药品说明书及标签药学相关信息撰写指导原则(试行)》 | 2023/3/21 |
| 21 | 2023 年第 21 号 | 《治疗卵巢癌新药临床研发技术指导原则(试行)》 | 2023/3/21 |
| 22 | 2023 年第 22 号 | 《儿童抗肿瘤药物临床研发技术指导原则》 | 2023/3/24 |
| 23 | 2023 年第 23 号 | 《化学药品创新药Ⅲ期临床试验前会议药学共性问题及相关技术要求(试行)》 | 2023/3/24 |
| 24 | 2023 年第 24 号 | 《生理药代动力学模型在儿科人群药物研发中应用的技术指导原则》 | 2023/3/28 |
| 25 | 2023 年第 25 号 | 《抗肿瘤抗体偶联药物临床研发技术指导原则》 | 2023/4/7 |
| 26 | 2023 年第 26 号 | 《基于动物法则的药物研究技术指导原则(试行)》 | 2023/4/7 |
| 27 | 2023 年第 27 号 | 《成人用药数据外推至儿科人群的定量方法学指导原则(试行)》 | 2023/4/12 |
| 28 | 2023 年第 28 号 | 《呼吸道胞病毒感染药物临床试验技术指导原则》 | 2023/4/12 |
| 29 | 2023 年第 29 号 | 《基因治疗血友病临床试验设计技术指导原则》 | 2023/4/14 |
| 30 | 2023 年第 30 号 | 《与恶性肿瘤治疗相关中药新药复方制剂临床研发技术指导原则(试行)》 | 2023/4/14 |
| 31 | 2023 年第 31 号 | 《慢性乙型肝炎病毒感染治疗药物临床试验技术指导原则》 | 2023/4/27 |
| 32 | 2023 年第 32 号 | 《肿瘤主动免疫治疗产品临床试验技术指导原则(试行)》 | 2023/4/26 |
| 33 | 2023 年第 33 号 | 《人源干细胞产品药学研究与评价技术指导原则(试行)》 | 2023/4/27 |
| 34 | 2023 年第 34 号 | 《抗肿瘤光动力治疗药物临床研发技术指导原则(试行)》 | 2023/4/28 |
| 35 | 2023 年第 35 号 | 《非阿片类术后镇痛新药临床试验设计技术指导原则》 | 2023/6/9 |
| 36 | 2023 年第 36 号 | 《新药获益-风险评估技术指导原则》 | 2023/6/25 |
| 37 | 2023 年第 37 号 | 《人源性干细胞及其衍生细胞治疗产品临床试验技术指导原则(试行)》 | 2023/6/21 |
| 38 | 2023 年第 39 号 | 《临床试验中的药物性肝损伤识别、处理及评价指导原则》 | 2023/7/10 |
| 39 | 2023 年第 40 号 | 《人乳头瘤病毒疫苗临床试验技术指导原则》 | 2023/7/11 |
| 40 | 2023 年第 41 号 | 《中药新药临床试验用药品的制备研究技术指导原则》 | 2023/7/25 |
| 41 | 2023 年第 42 号 | 《其他来源于古代经典名方的中药复方制剂药学研究技术指导原则(试行)》 | 2023/7/25 |
| 42 | 2023 年第 43 号 | 《罕见疾病药物开发中疾病自然史研究指导原则》 | 2023/7/27 |
| 43 | 2023 年第 44 号 | 《以患者为中心的药物临床试验设计技术指导原则(试行)》 | 2023/7/27 |
| 44 | 2023 年第 44 号 | 《以患者为中心的药物临床试验实施技术指导原则(试行)》 | 2023/7/27 |
| 45 | 2023 年第 44 号 | 《以患者为中心的药物获益-风险评估技术指导原则(试行)》 | 2023/7/27 |
| 46 | 2023 年第 45 号 | 《2 型糖尿病口服药物复方制剂研发指导原则》 | 2023/8/2 |
| 47 | 2023 年第 46 号 | 《抗体偶联药物非临床研究技术指导原则》 | 2023/9/27 |
| 48 | 2023 年第 47 号 | 《延缓慢性肾脏病进展的药物临床试验技术指导原则》 | 2023/9/28 |
| 49 | 2023 年第 48 号 | 《狼疮肾炎治疗药物临床试验技术指导原则》 | 2023/9/28 |
| 50 | 2023 年第 49 号 | 《多发性硬化治疗药物临床试验技术指导原则》 | 2023/9/28 |
| 51 | 2023 年第 50 号 | 《干眼治疗药物临床试验技术指导原则》 | 2023/9/28 |
| 52 | 2023 年第 53 号 | 《基于人用经验的中药复方制剂新药药学研究技术指导原则(试行)》 | 2023/10/18 |
| 53 | 2023 年第 54 号 | 《脂质体药物质量控制研究技术指导原则》 | 2023/10/19 |
| 54 | 2023 年第 54 号 | 《脂质体药物非临床药代动力学研究技术指导原则》 | 2023/10/19 |
| 55 | 2023 年第 55 号 | 《糖尿病视网膜病变相关中药新药临床研发技术指导原则(试行)》 | 2023/11/14 |
| 56 | 2023 年第 58 号 | 《特应性皮炎治疗药物临床试验技术指导原则》 | 2023/12/1 |
| 57 | 2023 年第 57 号 | 《氟[18F]脱氧葡糖注射液仿制药药学研究技术要求(试行)》 | 2023/12/1 |
| 58 | 2023 年第 59 号 | 《新药临床安全性评价技术指导原则》 | 2023/12/1 |
| 59 | 2023 年第 60 号 | 《细胞和基因治疗产品临床相关沟通交流技术指导原则》 | 2023/12/29 |
| 60 | 2023 年第 61 号 | 《人纤维蛋白原临床试验技术指导原则(修订版)》 | 2023/12/29 |
附件9 ICH 正在活跃的议题
ICH 正在活跃的议题
| 序号 | ICH 协调议题名称 |
|---|---|
| 1 | E2B(R3):临床安全数据的管理:个例安全报告传输的数据元素 |
| 2 | E2D(R1):上市后安全性数据的管理:快速报告的定义和标准 |
| 3 | E6(R3):药物临床试验质量管理规范(GCP) |
| 4 | 常设儿科(Standing Paediatric) |
| 5 | E11A:儿科外推 |
| 6 | E14/S7B:致QT/QTc 间期延长及潜在致心律失常作用的临床与非临床评价问答 |
| 7 | E20:适应性临床试验 |
| 8 | E21:临床试验纳入孕期和哺乳期妇女 |
| 9 | M1:监管活动医学词典 |
| 10 | M2 及M8 亚组:监管信息电子传输标准 |
| 11 | M4Q(R2):人用药品注册通用技术文档:质量 |
| 12 | M7(R3):评估和控制药物中的 DNA 活性(致突变)杂质以限制潜在的致癌风险 |
| 13 | M10:生物分析方法验证及样品分析 |
| 14 | M11:临床电子结构化协调方案(CeSHarP) |
| 15 | M12:药物相互作用研究 |
| 16 | M13:口服固体速释制剂的生物等效性 |
| 17 | M14:使用真实世界数据进行药品安全性评估的药物流行病学研究:规划和设计指导原则 |
| 18 | M15:模型引导的药物开发 |
| 19 | Q1/Q5C:稳定性系列指导原则修订 |
| 20 | Q2(R2)/Q14:分析过程开发和Q2(R1)分析方法验证的修订 |
| 21 | Q3E:杂质:对药品和生物制品的可提取物和可浸出物的评估和控制 |
| 22 | Q3C(R9):杂质:残留溶剂的指导原则 |
| 23 | Q3D(R3):元素杂质指导原则 |
| 24 | Q5A(R2):来源于人或动物细胞系生物技术产品的病毒安全性评价 |
| 25 | Q9(R1):质量风险管理 |
| 26 | Q12:药品生命周期管理的技术与法规考虑 |
| 27 | Q13:原料药和制剂的连续制造 |
| 28 | S1B(R1):致癌性研究 |
| 29 | S5(R4):人用药物生殖毒性检测 |
| 30 | GDG:正式仿制药讨论组 |
| 31 | QDG:正式质量讨论组 |
| 32 | CGTDG:细胞与基因治疗讨论组 |
附件 10 2023 年药审中心开展的培训
2023 年药审中心开展的培训
| 序号 | 培训时间 | 培训主题 | 培训形式 | 参加人数 |
|---|---|---|---|---|
| 1 | 2023 年 3 月31 日 | 药审中心举办药品注册研发沟通交流主题线上宣讲会 | 线上 | 1.4 万人 |
| 2 | 2023 年 4 月26 日 | 药审中心与药品长三角分中心联举办化学新药药学沟通交流培训会 | 线下 | 2000 人 |
| 3 | 2023 年 5 月24 日 | 药审中心与药品大湾区分中心联举办化学新药药学沟通交流培训会 | 线下 | 300 人 |
| 4 | 2023 年 5 月24 日 | 药审中心指导,药品审评检查长三角分中心举办“细胞和基因治疗产品临床研发技术指导原则主题”线上培训会 | 线上 | 0.6 万人 |
| 5 | 2023 年 5 月29 日-30 日 | 国家药监局药品注册司和药审中心指导,药品审评检查长三角分中心举办《中药注册管理专门规定》培训班(上海) | 线下 | 1300 人 |
| 6 | 2023 年 6 月12 日-13 日 | 国家药监局药品注册司和药审中心指导,药品审评检查大湾区分中心举办《中药注册管理专门规定》培训班(深圳) | 线下 | 1000 人 |
| 7 | 2023 年 6 月16 日 | 药审中心举办加快新药上市过程中的沟通交流主题线上宣讲会 | 线上 | 0.7 万人 |
| 8 | 2023 年 6 月20 日 | 药审中心举办药品注册受理基本要求和常见问题主题(线上)宣讲会 | 线上 | 1 万人 |
| 9 | 2023 年 8 月25 日、28 日 | 药审中心举办ICH 《E6(R3):药物临床试验质量管理规范》宣贯座谈会 | 线下 | 130 人 |
| 10 | 2023 年 9 月5-6 日 | 药审中心与辽宁、吉林、黑龙江三省药监局联举办“支持东北医药产业发展药品注册技术系列培训班”(化药专场) | 线下 | 450 人 |
| 11 | 2023 年 9 月14—15 日 | 药审中心与辽宁、吉林、黑龙江三省药监局联举办“支持东北医药产业发展药品注册技术系列培训班”(生物制品专场) | 线下 | 300 人 |
| 12 | 2023 年 9 月15 日 | 药审中心举办创新药临床研究与评价技术指导原则主题线上宣讲会 | 线上 | 1.2 万人 |
| 13 | 2023 年 9 月21-22 日 | 药审中心与辽宁、吉林、黑龙江三省药监局联举办“支持东北医药产业发展药品注册技术系列培训班”(中药专场) | 线下 | 430 人 |
| 14 | 2023 年 9 月27 日 | 药审中心举办京津冀抗肿瘤创新药技术指导原则培训班 | 线下 | 300 人 |
| 15 | 2023 年 10 月13 日 | 药审中心举办药品注册核查检验专题线上宣讲会 | 线上 | 1.8 万人 |
| 16 | 2023 年 10 月20 日 | 药审中心举办化学创新药主题线上宣讲会 | 线上 | 0.6 万人 |
| 17 | 2023 年 10 月25 日 | 药审中心举办儿童用药相关指导原则专题线上宣讲会 | 线上 | 1 万人 |
| 18 | 2023 年 11 月9 日 | 药审中心举办“新药非临床研究与评价”线上宣讲会 | 线上 | 1 万人 |
| 19 | 2023 年 11 月14 日 | 药审中心举办化学仿制药主题线上宣讲会 | 线上 | 1.1 万人 |
| 20 | 2023 年 11 月17 日 | 药审中心举办中药注册技术专题培训宣讲会 | 线上 | 0.55 万人 |
| 21 | 2023 年 12 月6 日 | 药审中心举办“新药临床试验期间药物警戒专题”线上宣讲会 | 线上 | 0.7 万人 |
| 22 | 2023 年 12 月8 日 | 药审中心与药品大湾区分中心联举办生物制品变更管理技术指导培训会 | 线下 | 600 人 |
| 23 | 2023 年 12 月20 日 | 药审中心举办“吸入制剂研究与评价专题”线上宣讲会 | 线上 | 0.5 万人 |
| 24 | 2023 年 12 月22 日 | 药审中心与药品长三角分中心联举办生物制品变更管理技术指导培训会 | 线下 | 1000 人 |














 浙公网安备33011002015279
浙公网安备33011002015279
 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论